
Evrysdi (risdiplam) is an oral medication indicated to treat spinal muscular atrophy (SMA) in patients, including adults, children and infants from birth.
The drug was developed by Genentech, a subsidiary of Roche, in partnership with the SMA Foundation and PTC Therapeutics, a US-based pharmaceutical company. It was developed using PTC Therapeutics’ proprietary splicing platform.
Recommended Buyers Guides
Evrysdi is available in a maximum dose of 5mg administered once daily, directly distributed to patients’ homes in the US by Accredo Health, a speciality pharmacy.
Regulatory approvals for Evrysdi
The US Food and Drug Administration (FDA) approved Evrysdi for the treatment of SMA in adults and children aged two months and older in August 2020, following the submission of a new drug application in September 2019.
In May 2022, the FDA extended the label for Evrysdi to include infants under two months with SMA.
The drug was approved by the European Commission in March 2021. The European Medicines Agency granted PRIME and orphan drug designations to risdiplam in December 2018 and February 2019, respectively.
Evrysdi was given orphan drug designation in January 2017 while fast-track designation was granted by the FDA in April 2017.
The drug is also approved in Japan, Australia, and Brazil for SMA treatment.
Spinal Muscular Atrophy causes and symptoms
SMA is a serious, progressively fatal neuromuscular disorder affecting around one in every 10,000 infants, the main genetic cause of child mortality.
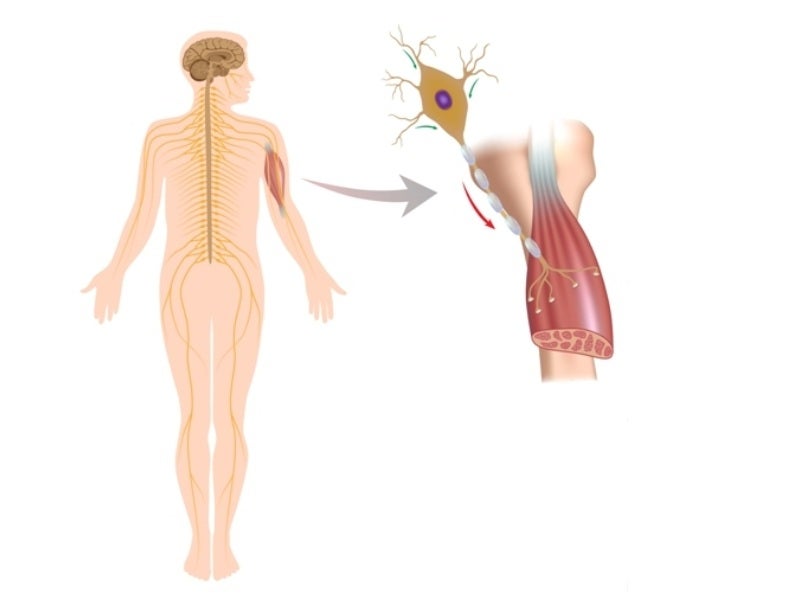
SMA is caused by a survival motor neuron 1 (SMN1) gene mutation, which results in SMN protein deficiency.
The protein is present in the body and is significant to the nervous system, which regulates muscles and movement. Nerve cells cannot function properly without the protein, leading to muscle fatigue.
SMA is classified into four main types 1, 2, 3, and 4, based on the disease’s intensity and the age when symptoms start.
SMA type 1 present between birth and six months of age is referred to as Werding Hoffman Disease, SMA type 2 present between six to 18 months of age is an intermediate form. Type 3 SMA is known as Kugleberg-Welander disease found in children over 18 months of age while type 4 SMA is seen in adults.
The physical strength of patients and their ability to walk, eat, or breathe may be diminished or destroyed, depending on the type of SMA.
The severity of the disease is also associated with the number of copies of the SMN2 gene in each individual.
Risdiplam’s mechanism of action
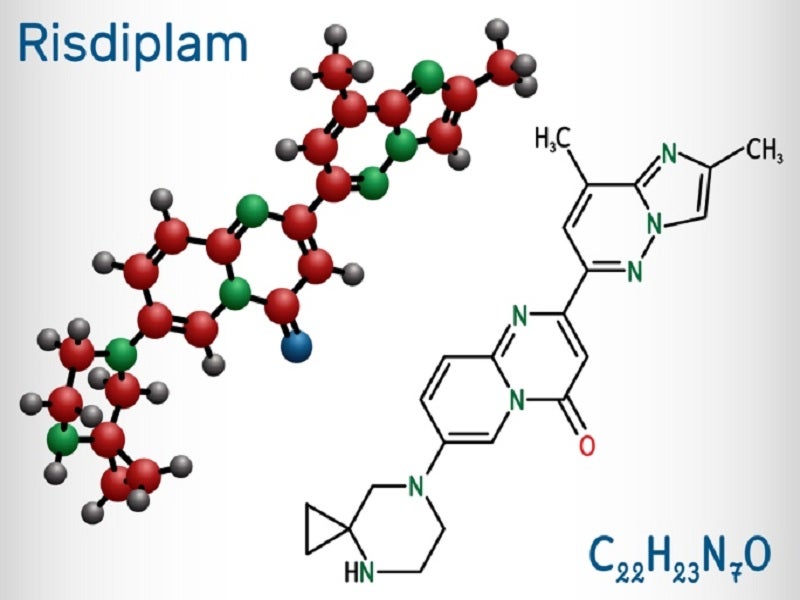
Risdiplam is a survival of SMN2 splicing modifier designed to treat patients with SMA caused by chromosome 5q mutations leading to SMN protein deficiencies.
The drug demonstrated a significant increase in exon 7 inclusion in SMN2 messenger ribonucleic acid transcripts and the production of full-length SMN protein in the brain during in vitro assays and studies in transgenic animal models of SMA.
In vitro and in vivo results suggest that risdiplam can induce alternate splicing of additional genes, including Forkhead box protein M1 (FOXM1) and MAP kinase activating death domain (MADD).
FOXM1 and MADD are believed to be active in the control of cell cycles and apoptosis, respectively, identified as potential contributors to adverse reactions seen in animals.
Clinical trials on Evrysdi
The FDA’s approval of Evrysdi was based on the results from two clinical studies, FIREFISH with infantile-onset SMA and SUNFISH with later-onset SMA.
FIREFISH (study one) was an open-label, multicentre, pivotal, and two-part clinical study that included 21 patients in the first stage and 41 patients in part two. The dose for part two was determined in part one.
SUNFISH (study two) was also a two-part, multicentre trial that enrolled 51 patients in part one and 180 patients in part two. Part one was an exploratory dose-finding study while part two was a randomised, double-blind, and placebo-controlled pivotal study.
Study one enrolled infants (aged between two and seven months) with type 1 SMA while study two included patients (aged from two to 25 years) with SMA types 2 and 3.
In the FIREFISH part one study, the efficacy was based on the survival without permanent ventilation and sitting without support for at least five seconds at 12 months of treatment measured by the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development Third Edition (BSID-III).
The BSID-III serves as a comprehensive formal assessment tool designed to identify developmental delays in early childhood.
Patients were administered the recommended Evrysdi dose of 0.2mg/kg a day. Approximately 41% of the patients were able to sit comfortably for roughly five seconds (BSID-III, gross motor scale) after 12 months of treatment.
After 12 months of Evrysdi treatment, 90% of the patients were alive without permanent ventilation and reached the age of 15 months or older.
After a minimum of 23 months of Evrysdi treatment, 81% of all patients (median age of 32 months) were alive without permanent ventilation.
The primary endpoint in the SUNFISH part two study was the mean change from baseline in the motor function measure (MFM-32) total score after 12 months of treatment with Evrysdi when compared to placebo.
Patients treated with Evrysdi reported a significantly greater change in motor function from baseline to placebo, as assessed by MFM-32.
At month 12, patients on Evrysdi saw an average 1.36 increase in their MFM-32 score, compared to a 0.19 decrease in placebo patients (inactive treatment).
The percentage of patients treated with Evrysdi who had a total score change of three or more in baseline MFM-32 was 38.3%, compared to 23.7% for placebo.
The treatment also improved upper limb motor function in children and adults compared to the baseline, as measured by the Revised Upper Limb Module, a secondary endpoint of the study.
Common adverse reactions observed were diarrhoea, fever, and rash in later-onset SMA (SUNFISH) and pneumonia, upper respiratory tract infection, vomiting, constipation, and cough, in infantile-onset SMA (FIREFISH).
The extended label approval of the drug for infants below the age of two was based on the RAINBOWFISH study conducted on newborns.
RAINBOWFISH evaluated the efficacy and safety of the drug in 25 babies with genetically diagnosed SMA, from birth to six weeks, with no displayed symptoms.
The findings indicated that presymptomatic infants undergoing Evrysdi treatment reached significant developmental milestones, including sitting and standing, with half of them achieving walking capabilities within 12 months of the treatment.
Notably, all infants were alive at the 12-month mark without the need for permanent ventilation.
Additional studies on Evrysdi
The JEWELFISH trial is an open-label exploratory study designed to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics in individuals aged six months to 60 years with SMA. Participants had previously undergone other investigational or approved SMA therapies for a minimum of 90 days before initiating Evrysdi.
MANATEE is a global phase II/III clinical trial designed to evaluate the safety and efficacy of GYM329, an anti-myostatin molecule that promotes muscle growth, in combination with Evrysdi for treating SMA in patients aged two to ten years.