
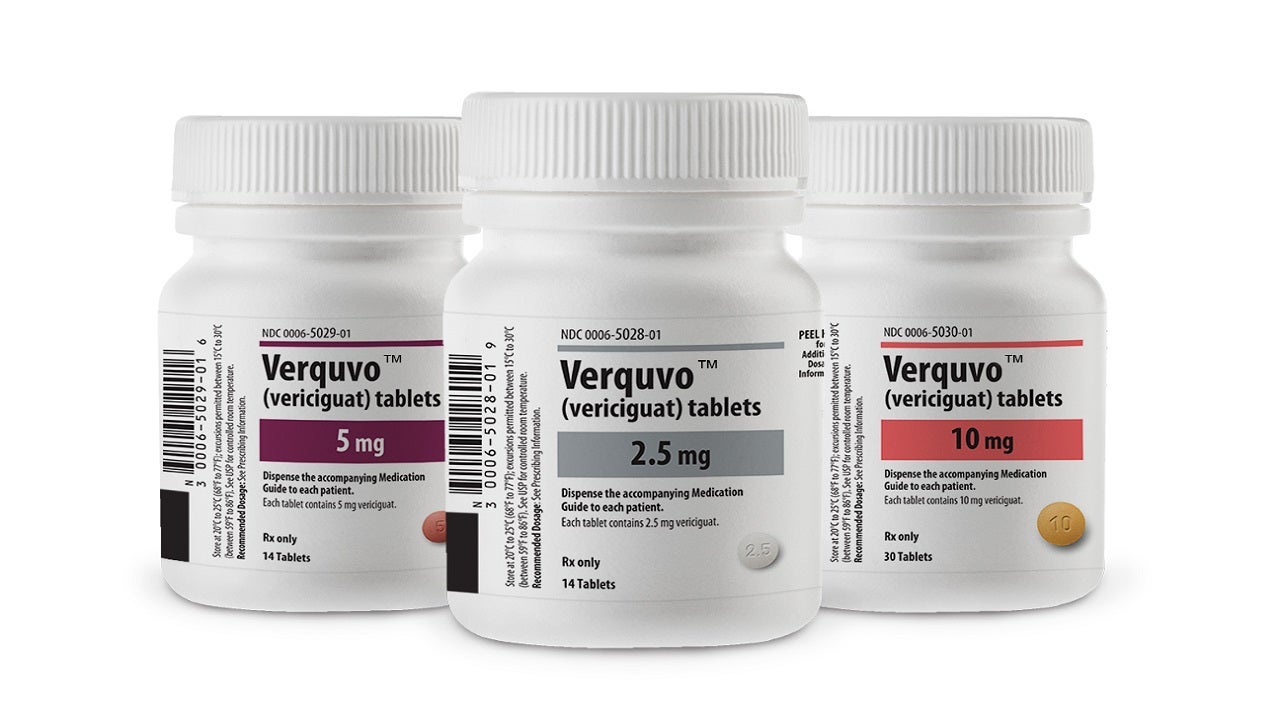
VERQUVO® (vericiguat) is a soluble guanylate cyclase (sGC) stimulator indicated to reduce the risk of cardiovascular death and hospitalisation due to heart failure (HF).
The drug is the first chronic HF treatment specifically approved for adult patients who are hospitalised for HF or have symptomatic chronic HF and an ejection fraction of less than 45%, requiring outpatient intravenous (IV) diuretics.
Recommended Buyers Guides
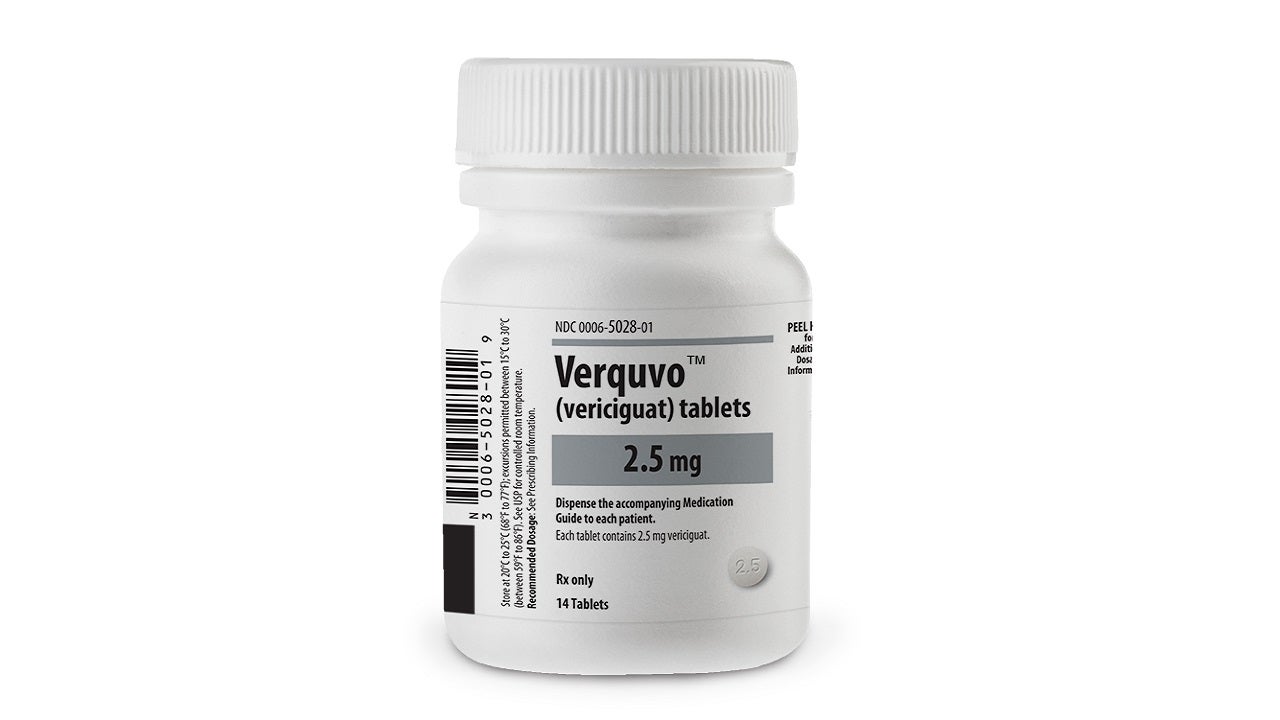
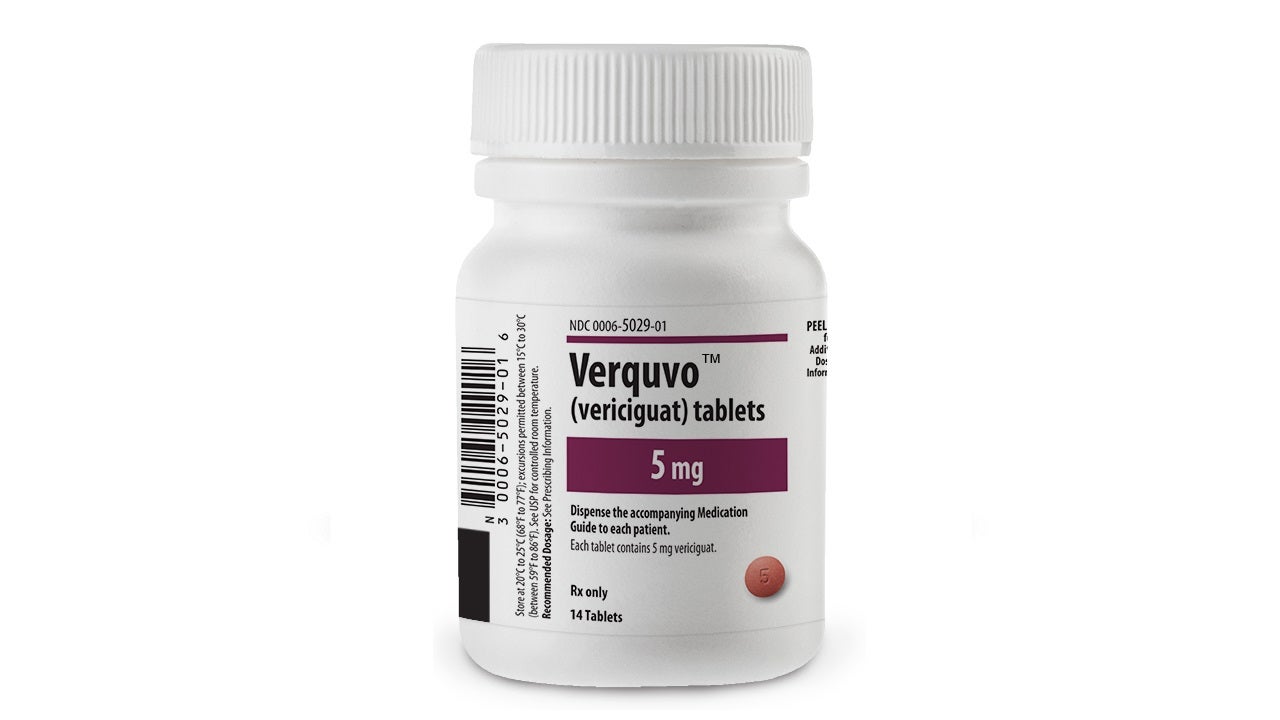
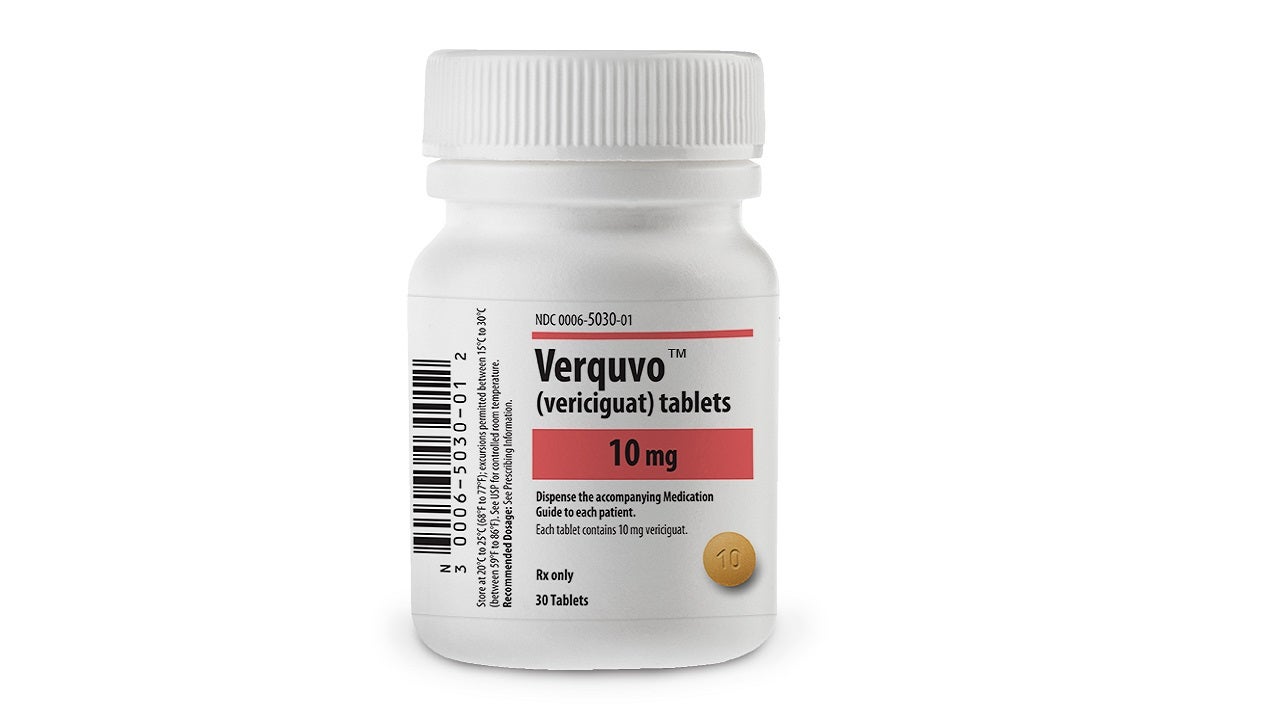
Co-developed by Bayer and Merck, VERQUVO is available as round, biconvex, film-coated tablets in 2.5mg (white-coloured), 5mg (brown-red coloured), and 10mg (yellow-orange coloured) dosage strengths for oral administration.
VERQUVO regulatory approvals
In May 2020, Bayer and Merck submitted a new drug application (NDA) for vericiguat to the US Food and Drug Administration (FDA). The NDA was accepted for priority review in July 2020.
In June 2020, the companies submitted a marketing authorisation application (MAA) for vericiguat in the European Union (EU) and Japan, followed by China in August 2020. MAAs for the drug have also been submitted in multiple other countries worldwide.
The US FDA approved VERQUVO for the treatment of heart failure with reduced ejection fraction (HfrEF) in January 2021.
In May 2021, the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) recommended vericiguat for marketing in the EU based on results from the pivotal Phase III VICTORIA trial, which were published in The New England Journal of Medicine (NEJM) in March 2020.
The data showed that vericiguat significantly reduced the combined risk of cardiovascular death or heart failure hospitalisations compared with background therapy alone, following a worsening heart failure event.
In June 2021, Japan’s Ministry of Health, Labour and Welfare (MHLW) approved the drug for the treatment of patients with chronic HF who are receiving standard treatment for the condition.
VERQUVO was also approved in Europe in July 2021 and in China in May 2022 based on the VICTORIA trial.
Heart failure with reduced ejection fraction causes and symptoms
HFrEF, also known as systolic heart failure, is characterised by the heart’s inability (particularly that of the left ventricle) to properly eject sufficient blood during the contraction phase.
Approximately 6.2 million patients in the US suffer from HF, around 40% to 50% of whom have HFrEF.
Around 30% of symptomatic chronic HF patients report a worsening of their condition, which is characterised by progressive signs or a recent HF case.
Approximately 50% of patients with deteriorating chronic HFrEF are re-hospitalised within 30 days of deterioration, and an estimated one in five patients with deteriorating chronic HFrEF die within two years.
The typical symptoms of HFrEF include weakness, oedema, orthopnoea, dyspnoea, and paroxysmal nocturnal dyspnoea.
The FDA approved VERQUVO for the treatment of heart failure with reduced ejection fraction (HfrEF) in January 2021.
VERQUVO mechanism of action
Vericiguat is an oral, once-daily sGC stimulator that acts as an essential enzyme in the nitric oxide (NO) signalling pathway.
When NO binds to sGC, the enzyme expedites the production of intracellular cyclic guanosine monophosphate (cGMP), a second messenger that plays a role in the management of vascular tone, cardiac contractility, and cardiac remodelling. HF is associated with impaired NO synthesis and decreased sGC activity, which can lead to myocardial and vascular dysfunction.
By explicitly activating sGC, independently and synergistically with NO, vericiguat increases intracellular cGMP levels, causing smooth muscle relaxation and vasodilation.
Clinical trials on VERQUVO
The FDA’s approval of VERQUVO was based on results from a randomised, double-blind, parallel-group, placebo-controlled, event-driven, multi-centre, pivotal Phase III clinical trial named VICTORIA.
A total of 5,050 adult patients with symptomatic chronic HF were randomised to receive either 10mg of VERCUVO or corresponding placebo.
The treatment began with a 2.5mg dosage strength once daily, which was then increased to 5mg at approximately two-week intervals followed by a target dose of up to 10mg as tolerated. The doses of placebo were adjusted similarly.
Approximately one year later, 90% of patients in both the VERQUVO and placebo arms were treated with the 10mg target maintenance dose.
The study’s primary goal was to measure the time to the first cardiovascular death event or HF hospitalisation case. The median follow-up period was 11 months for the primary outcome.
The mean left ventricular ejection fraction (LVEF) of patients who participated in the study was 29%. Around half of all patients had an EF of less than 30%, while 14% of the group had an EF between 40 and 45%.
At randomisation, the median NT-pro B-type natriuretic peptide (NT-proBNP) level was 2,800pg/ml.
In the clinical trial, VERQUVO was better than the placebo in reducing the risk of cardiovascular death or HF, based on a time-to-event evaluation.
A 4.2% reduction was observed in the annualised absolute risk (ARR) with VERQUVO compared with placebo during the study. The treatment result represented a decline in both cardiovascular death and HF.
The most common adverse reactions observed in the patients during the clinical trial were anaemia and hypotension.
In addition, Bayer and Merck have comprehensively committed to dedicated investment in a full umbrella clinical research programme named DISCOVERI. The DISCOVERI programme aims to expand understanding of vericiguat’s clinical utility while exploring its use in heart failure patients beyond the criteria set in VICTORIA.