
Sarepta Therapeutics has announced its plans to submit a New Drug Application (NDA) for accelerated approval of its drug golodirsen (SRP-4053), a treatment for the neuromuscular disorder Duchenne Muscular Dystrophy (DMD).
Golodirsen is a phosphordiamidate morpholino oligimer specifically designed to treat patients with genetic mutations subject to skipping exon 53 of the DMD gene.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
The announcement follows Sarepta’s recent receipt of the final minutes from its Type C meeting with the US Food and Drug Administration’s (FDA) Division of Neurology Products, held in February 2018 to discuss the regulatory pathway for golodirsen.
“Sarepta is thankful for the FDA Neurology Division’s thoughtful and direct guidance regarding golodirsen,” Sarepta CEO Doug Ingram said.
“Obviously, whether golodirsen will obtain accelerated approval is a review decision that will come after the submission and review of our NDA. But we greatly appreciate the willingness of the Neurology Division to engage and provide clear direction to us on the steps necessary to support an NDA submission for accelerated approval.”
US-based Sarepta is known for its research into and development of precision genetic medicine to treat rare neuromuscular diseases. Its current focus is on the advancement of its potentially disease-modifying drug candidates for DMD, one of the most common fatal genetic disorders. DMD is seen in approximately one in every 3,500 – 5,000 male births worldwide, causing progressive muscle loss and premature death.
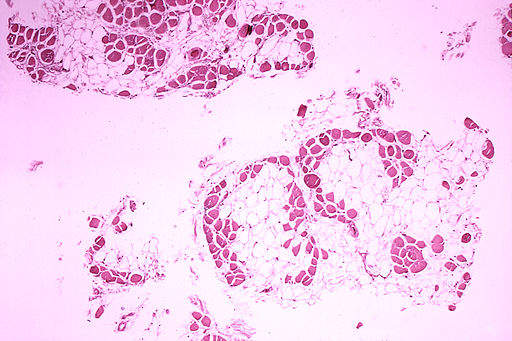
A Phase I/II study assessing the efficacy and safety of golodirsen was conducted in the third quarter of 2017. In total 25 boys with a genetic mutation of the DMD gene were enrolled in the study, with the drug showing statistically significant results on all biological endpoints, including properly exon-skipped RNA transcript using reverse transcription polymerase chain reaction, quantity of dystrophin expression using western blot and dystrophin intensity pursuant to immunohistochemistry.
These positive findings, in addition to the FDA’s recent feedback, have led Sarepta to initiate plans for a rolling submission of a golodirsen NDA by the end of 2018.
The success of Sarepta’s request for accelerated approval will partially rely on its provision of substantial evidence of golodirsen’s effect on dystrophin from a single study. Sarepta has proposed that its Study 4045-301 (ESSENCE), a Phase III trial assessing the efficacy of golodirsen and casimersen, serve as the post-marketing confirmatory study. The Neurology Division agreed to ESSENCE acting as a confirmatory study providing Sarepta gives details on how it will enroll and complete the study in light of an accelerated approval.
In addition, the complete submission must include long-term animal toxicology studies, which will be completed in the fourth quarter of 2018. As such, Sarepta has said it anticipates the NDA submission to be completed in late 2018.
Sarepta’s request has come under scrutiny due to the controversy surrounding its first Duchenne treatment, Exondys 51. Research into the drug showed it made 0.28% of the normal amount of the protein dystrophin, which is missing in DMD patients. Despite protests from some FDA members who said the drug was unlikely to make a significant difference to patients, the treatment was approved in 2016. By comparison, golodirsen has demonstrated a 1.2% increase in dystrophin, 11 times the amount found in DMD patients and more than triple the amount in Exondys.
