Consulting and Regulatory Services


Drug development is very expensive and carries important risks. Success in this area can be measured by a rewarding return on investment as a result of a broad marketing authorisation with an attractive indication and a highly competitive marketing positioning.
This calls for a clever development plan and regulatory strategy based on a cautious, but robust gap and Strengths, Weaknesses, Opportunities, Threats ( SWOT) analysis, which also account for the pertinent regulatory guidelines and how these are implemented (as can be learnt from recent public assessment reports).
It is important to agree on the best suited regulatory procedure from options such as centralised procedure (CP), decentralised procedure (DCP) and mutual recognition procedure ( MRP) for the documentation needed for different regulatory cases, including full and mixed-full according to Article 8(3), generic according to Article 10(1), bridged hybrid according to Article 10(3), biosimilar according to Artical 10(4) and well-established-use according to Artical 10(a).
In some cases, regulatory scientific advice (SA) may help, while in many cases it will not. Even if non-binding, SA sets requirements that become part of the product’s dossier with a possible impact on all further regulatory dealings.
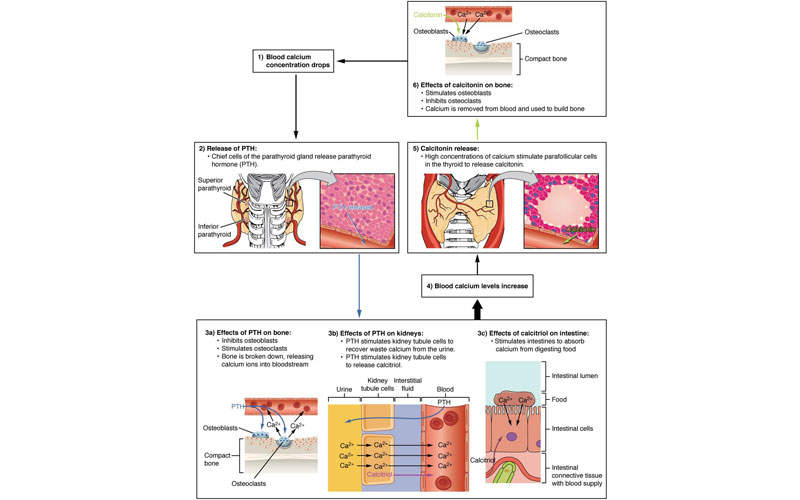
The clinical development plan ought to be a direct reflection of the regulatory strategy with the purpose to provide adequate and sufficient evidence of the drug’s efficacy and safety such that the medication can be authorised as a tangible while worthwhile extension of the options for medical care.
Good planning involves aspects of both content (scientific excellence) and format (regulatory compliance).
ACPS are experts in aligning these needs in time- and cost-efficient fashion.