
Anthim (obiltoxaximab) is an injectable drug developed by Elusys Therapeutics for the treatment of inhalational anthrax caused by bacillus anthracis.
Elusys Therapeutics submitted the biological license application (BLA) for the drug to the US Food and Drug Administration (FDA) on 20 March 2015. The BLA was accepted by the FDA and the drug was granted orphan drug designation in the US.
Recommended White Papers
Recommended Buyers Guides
The US FDA approved Anthim (obiltoxaximab) on 21 March 2016 as a treatment for inhalational anthrax in adults and children in combination with antibacterial drugs.
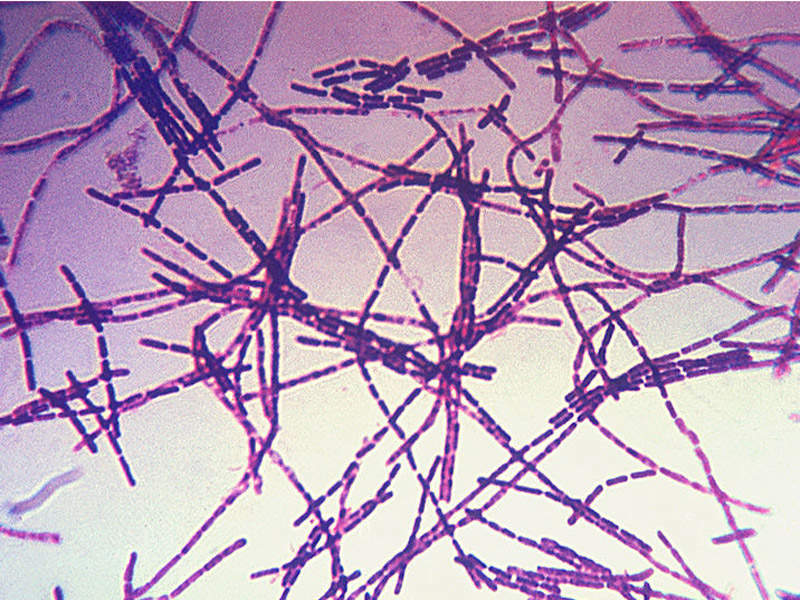
Anthrax causes and types
Anthrax is a chronic bacterial infection caused by gram positive, rod-shaped bacteria known as bacillus anthracis, which usually affects domestic and wild animals. There are different types of the illness namely cutaneous, inhalational, gastrointestinal and injection.
Inhalational anthrax is the deadliest form and usually occurs when a person inhales spores present in the air, entering the body and infecting the lungs.
The symptoms usually associated with the disease are fever, chills, chest discomfort, shortness of breath, dizziness, cough, nausea, stomach pain, headache, sweats, extreme tiredness and body aches.
Inhalation anthrax is often diagnosed by chest X-ray or CT scans in suspected patients.
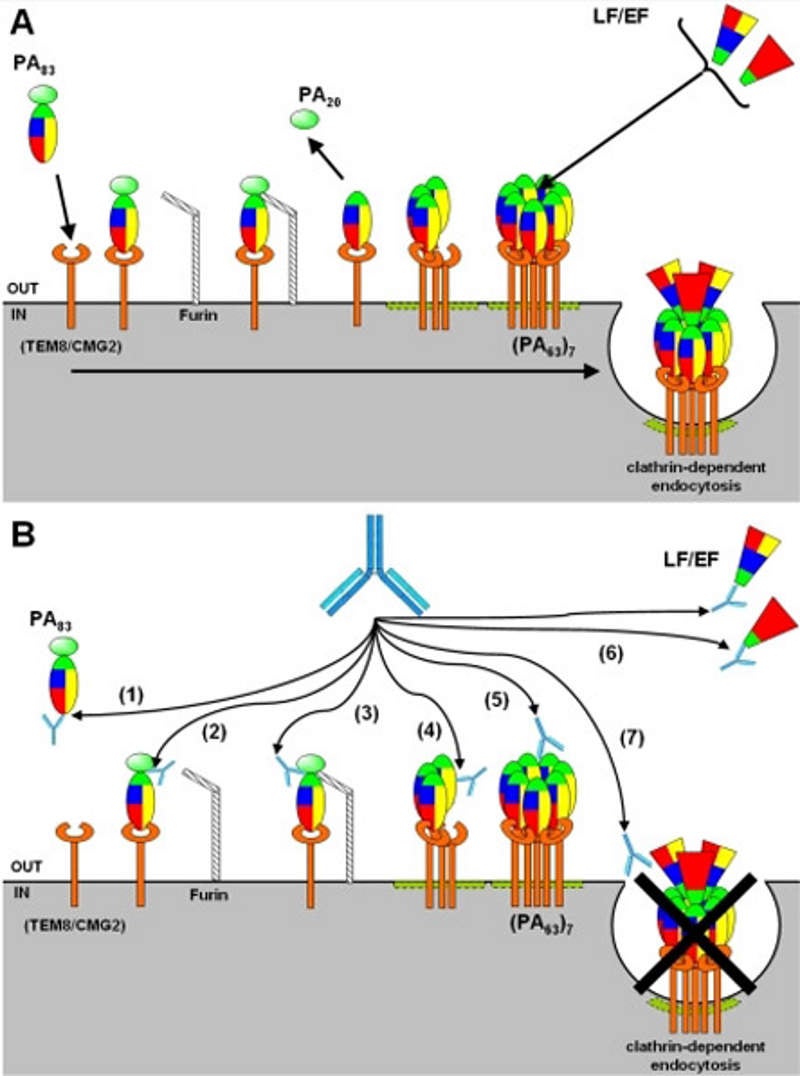
Anthim’s mechanism of action
Anthim (obiltoxaximab) is a high-affinity humanised and deimmunised monoclonal antibody that binds to the protective antigen component of anthrax toxin.
Anthim’s toxin neutralising activity helps in preventing the entry of anthrax toxins produced by bacillus anthracis into the susceptible cells. The pathogenicity of the bacteria depends upon the two types of toxins namely lethal toxins (LT) and edema toxin (ET).
The drug prevents the toxins entry thereby avoiding the spreading of the toxin throughout the body.
Clinical trials on obiltoxaximab
The US FDA’s approval of Anthim (obiltoxaximab) was based on the results obtained from three phase 3 clinical trials conducted to evaluate the safety and efficacy of the single intravenous dose (16mg/kg) of obiltoxaximab in healthy adult men and women aged above 18 years.
The first clinical trial was double-blind, randomised, placebo-controlled study, which enrolled 280 healthy adult subjects to demonstrate the safety and tolerability of repeat IV administration of obiltoxaximab (16mg/kg).
The subjects were randomised to receive obiltoxaxim (210) and placebo (70). The adverse events reported during the trail were mild to moderate, whereas the most frequently reported AEs in subjects randomised to obiltoxaximab were pruritus, headache, rash and urticaria.
The second trial was a double-blind, randomised and placebo-controlled study, which enrolled 70 healthy volunteers to demonstrate the safety and tolerability of repeat IV administration of obiltoxaximab (16mg/kg).
The subjects were classified into two treatment groups Group A (35 Subjects) and Group B (35 subjects). Group A received Obiltoxaximab on days 1 and 14 and placebo on days 120 of the study; Group B received Obiltoxaximab on day 1 and 120 and placebo on day 14 of the study. The most adverse events reported during the study were infusion site swelling, erythema and pain.
The third trial was an open-label, randomised, parallel group drug-drug interaction study to evaluate the safety and efficacy of obiltoxaximab when given along with ciprofloxacin. The study enrolled 40 healthy volunteers between 18 to 65 years. The subjects were placed into two treatment groups of 20 each.
Group 1 received Obiltoxaximab (16mg/kg) and single dose IV ciprofloxacin (400mg) followed by an oral ciprofloxacin (750mg), whereas Group 2 received IV obiltoxaximab (16mg/kg) alone. The only adverse event reported during the trial was urticaria.