
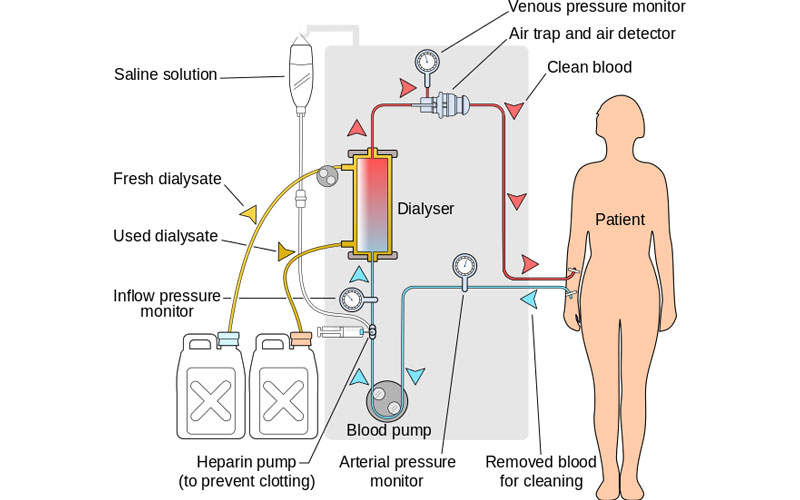
Parsabiv™ (etelcalcetide) is an intravenous injectable calcimimetic agent developed by Amgen for the treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD) that were previously treated with haemodialysis.
Amgen submitted a new drug application (NDA) for Parsabiv™ to the US Food and Drug Administration (FDA) in August 2015. The FDA accepted the NDA for review in November 2015, and set a Prescription Drug User Fee Act (PDUFA) target action date of 24 August 2016.
Recommended White Papers
Recommended Buyers Guides
Formerly known as AMG416, Parsabiv™ was approved by the FDA for the treatment of SHPT in December 2017. It is one of the first calcimimetic agents to have become available in the US for intravenous administration.
Amgen also submitted a marketing authorisation application (MAA) for the drug to the European Medicines Agency (EMA) in September 2015. The European Commission (EC) approved Parsabiv™ for the treatment of SHPT in November 2016.
Secondary hyperparathyroidism
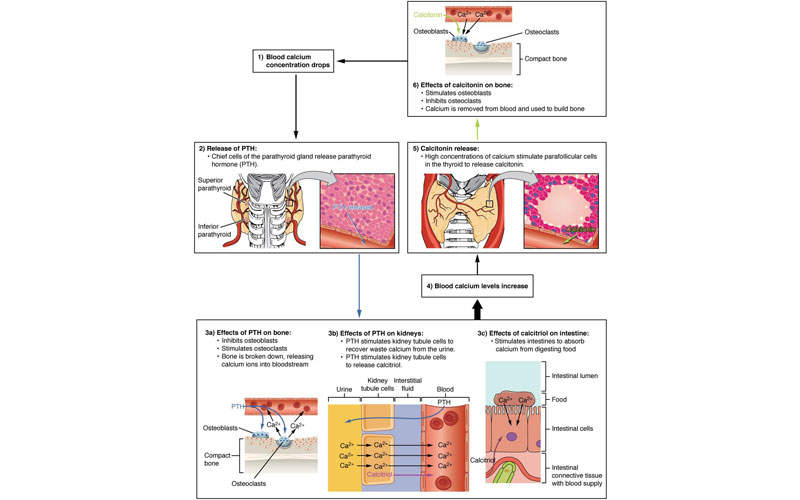
SHPT is a chronic disease associated with the parathyroid gland and is characterised by excessive secretion of the parathyroid hormone (PTH) in response to hypocalcaemia (low blood calcium levels). The disorder often develops in CKD patients.
It occurs in 90% of patients with chronic renal failure on haemodialysis, and is usually referred to as renal hyperparathyroidism.
Hyperparathyroidism affects approximately two million people worldwide, including around 350,000 people in Europe and 450,000 people in the US.
Parsabiv’s mechanism of action
Parsabiv™ is an intravenously administered calcimimetic agent. The drug binds to the calcium-sensing receptors and activates them on the parathyroid gland, decreasing PTH secretion. The drug is administered intravenously after every dialysis session.
Clinical trials
The FDA’s NDA approval for Parsabiv™ was based on three Phase III randomised, placebo-controlled, double blind and double dummy clinical trials.
The head-to-head Phase III clinical study was conducted for 26 weeks on 683 patients, and evaluated etelcalcetide’s efficiency compared to cinacalcet.
The patients randomised with Parsabiv™ received intravenous doses of etelcalceitide three times a week at the end of each dialysis session, and daily oral doses of placebo tablets.
The patients who were randomised with cinacalcet received daily oral doses of cinacalcet tablets and doses of placebo three times a week, at the end of each dialysis session.
The primary endpoint of the study was the proportion of patients with a greater than 30% reduction from baseline in mean PTH levels during weeks 20 and 27. The trial achieved the primary end point of groups of patients with a greater than 30% reduction in mean PTH from baseline.
It also achieved the secondary end points, including greater than 50% reductions from baseline PTH during the efficacy assessment phase (EAP), and the mean number of days of vomiting or nausea a week in the first eight weeks.
Presented at American Society of Nephrology (ASN), the study’s results reported 92.9% treatment emergent adverse events (TEAE) in patients with Parsabiv™, and 90% in patients with cinacalcet.
The TEAEs reported in at least 10% of the patients treated with Parsabiv™ or cinacalcet included decreased blood calcium, nausea, vomiting, diarrhoea, hypocalcaemia, and cardiac failure.
Serious adverse events were reported in both the etelcalcetide (25.1%) and cinacalcet (27.3%) groups, while fatal adverse events were reported in 2.7% of the etelcalcetide group and 1.8% of the cinacalcet group.
The EC’s approval was also based on the results obtained from three Phase III clinical studies, which each met their primary endpoints. The clinical studies also included two pooled placebo-controlled trials that evaluated more than 1,000 patients in a head-to-head study comparing Parsabiv™ with cinacalcet.