
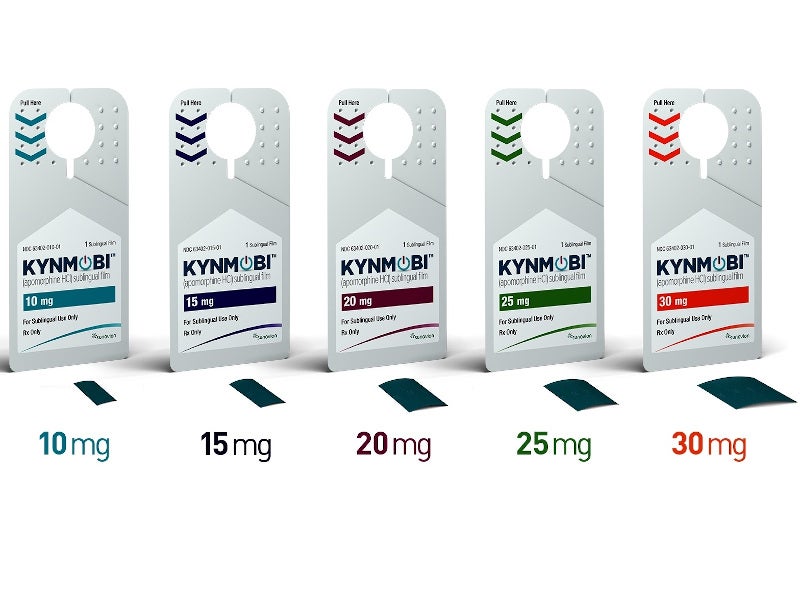

KYNMOBI® (APL-130277) is a sublingual formulation of apomorphine approved for the treatment of OFF episodes in Parkinson’s disease.
The drug was developed by Sunovion Pharmaceuticals, a wholly owned subsidiary of Sumitomo Pharma (previously Sumitomo Dainippon Pharma).
Recommended Buyers Guides
KYNMOBI is available as a sublingual film coated with apomorphine, which can be used up to five times a day at doses of between 10mg and 30mg. The drug is administered non-invasively by placing it under the tongue, where it is dissolved.
KYNMOBI is currently in Phase III of clinical development in Europe. Sunovion entered an agreement with pharmaceutical company BIAL in September 2021, granting the commercial license rights for APL-130277 in Europe.
Under the terms of the agreement, BIAL will be responsible for submitting the marketing applications for APL-130277 in Europe and will own the commercial rights to distribute and commercialise the drug in the European Union (EU), the European Economic Area (EEA) and the UK.
Sunovion received an upfront payment and is entitled to receive milestone payments as part of the agreement. It retains the commercial rights to market KYNMOBI in North America and all other regions except the EU, the EEA and the UK.
Regulatory approvals on KYNMOBI
In March 2018, Sunovion submitted a new drug application (NDA) for KYNMOBI to the US Food and Drug Administration (FDA). This was accepted for review in June 2018.
In January 2019, Sunovion received a complete response letter (CRL) from the FDA requesting additional data. The company resubmitted the NDA to the FDA in November 2019 by including details of the packaging and additional analysis of clinical data.
The FDA approved KYNMOBI for the treatment of short-term, intermittent OFF episodes in people with Parkinson’s disease in May 2020.
In June 2020, Health Canada approved KYNMOBI for the acute and intermittent treatment of OFF episodes in Parkinson’s disease patients.
Causes and symptoms of OFF episodes in Parkinson’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that occurs when nerve cells, or neurons, are damaged or die in a brain field that regulates movement. These neurons normally produce a chemical in the brain known as dopamine in the substantia nigra. If the neurons are damaged or become dysfunctional, less dopamine is produced leading to kinetic or movement issues.
The disease induces shaking, trembling, and difficulty in walking, standing and coordinating. symptoms of the disease usually start gradually and become more severe over time. Patients may have difficulty in walking and talking and experience significant changes in behaviour, in addition to sleep problems, depression, impaired memory and tiredness.
By 2030, around 1.2 million people in the US and ten million people worldwide are expected to be living with PD.
OFF episodes in PD are re-emergences or deteriorations of motor and non-motor symptoms normally controlled by oral levodopa and carbidopa. These episodes normally occur in the morning after waking up and during the day, and are experienced by 60% of people with PD within four to six years of diagnosis.
Symptoms of OFF episodes may include tremors, rigidity, nausea, vomiting, sleepiness, dizziness, oral mouth irritation such as redness, ulceration, mouth dryness, pain and swelling, hallucinations and slow motion, bowing-down posture, balance disturbance, communication problems, anxiety, and symptoms of depression.
KYNMOBI’s mechanism of action
KYNMOBI is a non-ergoline agonist of dopamine with a strong in-vitro binding affinity to the dopamine D4 receptor and intermediate affinity to the dopamine D2, D3 and D5 receptors, as well as the adrenergic dopamine α1D, α2B, α2C receptors.
The drug’s exact mechanism of action as a therapy for OFF episodes of Parkinson’s disease is unknown, although it is assumed to work by activating post-synaptic D2-type dopamine receptors within the brain’s caudate-putamen.
Clinical trials on KYNMOBI
The FDA’s approval of KYNMOBI was based on a Phase III randomised, double-blind, placebo-controlled, parallel-group, clinical trial named CTH-300, which was conducted in patients experiencing OFF episodes within PD.
The study included patients suffering from PD for a median duration of nine years and in the Hoehn and Yahr Phase III or less. Patients received a steady dosage of concomitant levodopa for at least four weeks prior to screening.
A total of 109 patients were randomised in a 1:1 ratio to receive either 10mg of KYNMOBI or placebo. The study included a titration phase and a maintenance phase of 12 weeks.
The titration process was subsequently increased to a maximal dosage of 35mg of KYNMOBI or until the full ‘on’ was achieved, as decided by the investigator and the patient. The administration of dose was allowed up to five times a day in the maintenance phase.
The study’s primary endpoint was the mean shift from pre-dose to 30 minutes post-dose in Movement Disorder Society-Unified Parkinson’s Disease Rating Scale, Part III (MDS-UPDRS III) score during the maintenance phase.
The KYNMOBI treatment group exhibited a minimum-square mean improvement of -11.1 points compared with -3.5 points for the placebo group. In addition, a significantly higher percentage of people administered with KYNMOBI had a patient-rated full ON response within 30 minutes at week 12, compared with placebo-treated patients.
Common adverse reactions observed during the study were dizziness, oropharyngeal reactions, nausea, and somnolence.
KYNMOBI is also being evaluated in an ongoing open-label, long-term study (CTH-301) to evaluate its safety and tolerability. Ad-hoc analysis from the trial announced in September 2021 showed that most patients experienced mild nausea and were able to continue treatment even in the absence of antiemetic treatment.
The analysis also showed that the MDS-UPDRS III score was greater in European patients than in North American patients. The MDS-UPDRS III score was -28.5 for European patients and -20.1 for North American patients at week 24, while this stood at -28.2 for European patients and -19.6 for North American patients at week 36.
In addition, a patient-rated full ON response within 30 minutes of dosing was observed by more than 75% of patients through 48 weeks in both regions.