
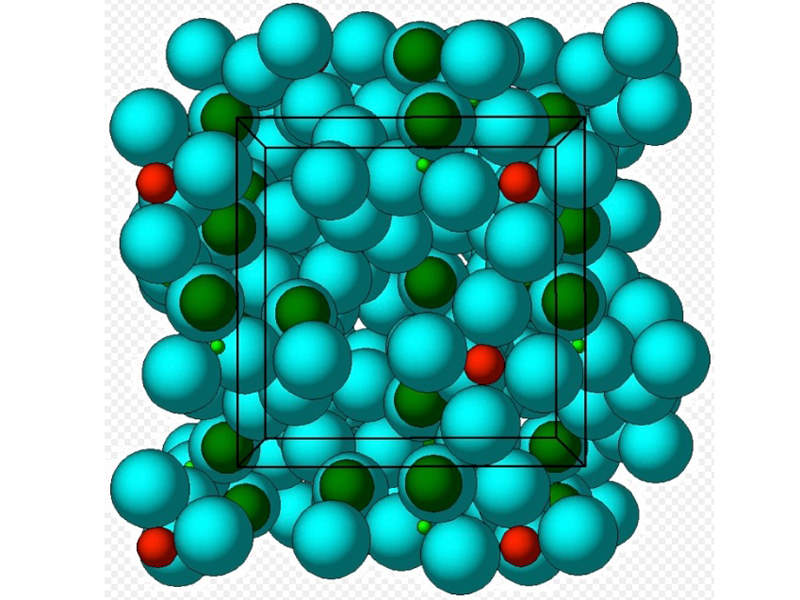
Lokelma is an oral suspension of sodium zirconium cyclosilicate that is indicated for the treatment of hyperkalaemia.
Formerly known as ZS-9, the drug was discovered and developed by global biopharmaceutical company AstraZeneca.
Recommended White Papers
Recommended Buyers Guides
A marketing authorisation application (MAA) for the drug was submitted to the European Medicines Agency (EMA) in December 2015. It received marketing authorisation from the European Commission (EC) in March 2018.
The US Food and Drug Administration (FDA) accepted a new drug application (NDA) for Lokelma in October 2016, and granted approval in May 2018.
Hyperkalaemia causes and symptoms
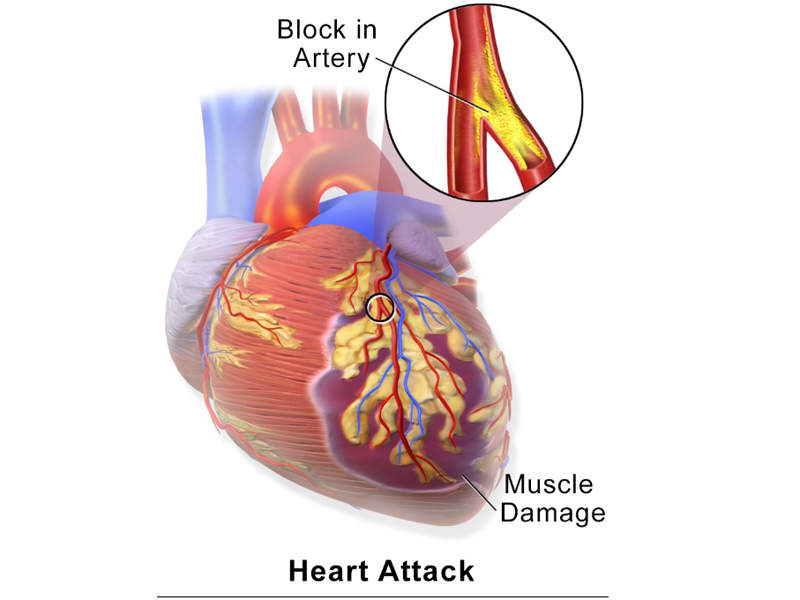
Hyperkalaemia is a life-threatening condition caused by high levels of potassium in the blood. The disease occurs due to reduced renal excretion and can lead to cardiac arrest by suppressing the electrical activity of the heart.
Hyperkalaemia is more risky for people with chronic kidney disease and for patients that take common medications for heart failure, such as renin-angiotensin-aldosterone system (RAAS) inhibitors.
Symptoms of hyperkalaemia are non-specific and are mostly associated with muscular or cardiac dysfunction. People with high potassium levels may experience symptoms such as muscle weakness, numbness, tingling and nausea.
Lokelma’s mechanism of action
Sodium zirconium cyclosilicate contained in Lokelma is an insoluble, non-absorbable inorganic crystalline compound that works as a potassium blinder. It attaches to potassium in the patient’s food and body fluids in the lumen of the gastrointestinal tract and lowers the serum potassium levels.
Lokelma is available in 5g and 10g packets.
Clinical trials on Lokelma
The US FDA’s approval for Lokelma was based on data from three double-blind, placebo-controlled trials and two open-label trials.
In the first clinical study, 753 patients with a median age of 66 years were randomised to 1.25g, 2.5g, 5g, or 10g of Lokelma three times daily or placebo. More than 60% of the randomised patients had chronic kidney disease, while 10% had heart failure, 62% had diabetes mellitus, and 67% were on RAAS inhibitor therapy.
The primary endpoint of the study was a significant reduction in serum potassium levels during the initial 48 hours of drug administration. The study met this primary endpoint for the 2.5g, 5g and 10g doses when compared to placebo.
The patients, who attained potassium level (mean serum potassium change) of 3.5-5mEq/l after taking Lokelma during the acute phase, were randomised again to 1.25g, 2.5g, 5g, or 10g of once-daily LOKELMA or once-daily placebo for 12 days. The primary endpoint was attained at 5g and 10g doses compared with placebo-randomised patient groups.
The second study was an open-label acute phase trial that enroled 2,258 patients with a median potassium level range of 5.6mEq/l. The patients were randomised to receive 10g of Lokelma three times daily with meals. The trial was followed by a double-blind, randomised withdrawal phase, in which patients that achieved potassium levels between 3.5 and 5mEq/l were randomised to one of the three doses of Lokelma or placebo once-daily for 28 days.
The primary endpoint of the study was the maintenance of mean serum potassium value change compared to placebo. Lokelma was able to maintain mean serum potassium levels lower than placebo.
Lokelma was also evaluated in an open-label extension trial, which saw patients that completed the 28-day randomisation continuing the treatment with Lokelma for more than 11 months. The study determined whether the serum potassium levels were maintained during continued therapy.
In another open-label 12-month study, Lokelma was evaluated in 751 hyperkalaemic patients whose baseline potassium level was 5.6mEq/l. To continue the maintenance of the potassium levels, Lokelma was administered 5g once daily and the dosage was increased to a maximum of 15g with a 5g increase in dose on every alternate day of the treatment, based on serum potassium level.
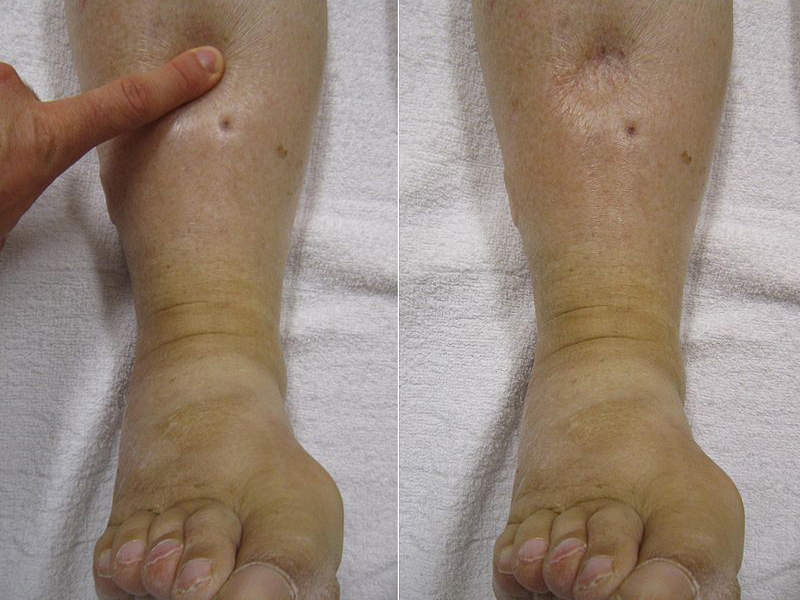
The most common adverse reactions of the drug were oedema-related events.