
LuxturnaTM (voretigene neparvovec) is approved for the treatment of patients with biallelic RPE65 mutation-associated retinal dystrophy.
The adeno-associated virus (AAV) vector gene therapy was discovered and developed by Spark Therapeutics.
Recommended White Papers
Recommended Buyers Guides
A biologics license application (BLA) for Luxturna was accepted for review by the US Food and Drug Administration (FDA) in July 2017 and granted priority review. It was also granted orphan drug and breakthrough therapy designations by the FDA.
Luxturna was approved by the FDA in December 2017. The drug is currently under review for marketing authorisation approval (MAA) by the European Medicines Agency (EMA) and has received orphan product designation in Europe.
The drug will be produced at Spark Therapeutics’ manufacturing facility in West Philadelphia, US. It is expected to be launched in the US market in Q1 2018.
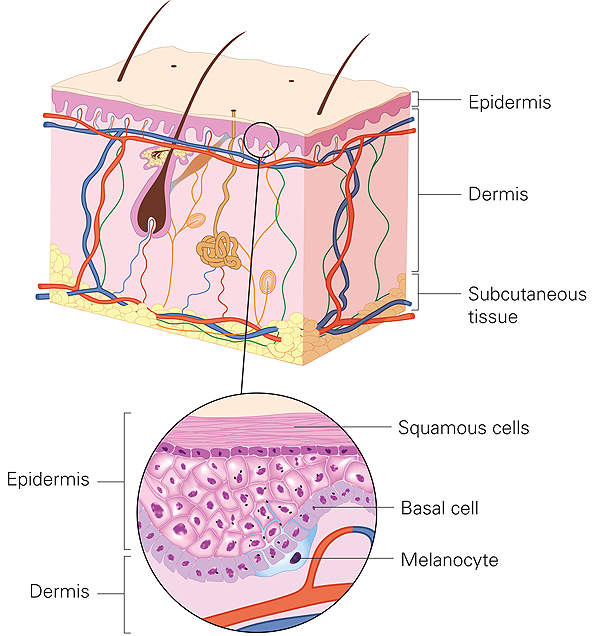
RPE65 mutation-associated inherited retinal disease causes
Also known as inherited retinal dystrophy, inherited retinal disease (IRD) is a rare blinding condition caused by mutation of biallelic RPE65 genes and frequently affects children and young adults.
The people affected by the disease may experience night blindness and involuntary eye movements in childhood or early adulthood. The disease may also result in peripheral, central or complete vision loss.
IRD is estimated to affect approximately 6,000 people across the US, Europe, the Americas and the Asia Pacific (APAC).
Luxturna’s mechanism of action
Luxturna contains a gene therapy designed to treat mutations in the RPE65 gene. The drug uses a neutralised virus as a vector to carry a functional gene into the affected eye tissue, enabling the body to produce any proteins missing due to genetic mutation.
The drug is available in a single dose vial and can be administered as sub-retinal injection.
Clinical trials on Luxturna
FDA approval for Luxturna was based on results obtained from two Phase I and one Phase III clinical studies. The Phase I trials conducted between 2007 and 2012 were open-label, dose-exploration safety studies, while the Phase III study conducted between 2013 and 2015 was an open-label, randomised and controlled efficacy study.
The clinical studies enrolled patients with biallelic RPE65 mutation-associated retinal dystrophy and sufficient viable retinal cells.
The Phase III clinical study enrolled 31 patients. Of these, 21 were administered with Luxturna sub-retinal injection, while the rest were randomised to the control group. One patient from each group discontinued from the study. All nine patients of the control group were administered with Luxturna after one year of observation.
The efficacy of Luxturna was evaluated using the multi-luminance mobility test (MLMT) score change from baseline to one year. The study was designed to find the changes in functional vision at seven different levels of illumination, ranging from 400lux to 1lux.
Other clinical outcomes of the study included white light full-field light sensitivity threshold (FST) testing and visual acuity.
The study results registered a statistically major difference between the Luxturna intervention group and control group participants in median bilateral MLMT score change and median first-treated eye MLMT score change after one year.
The study also showed that the change in visual acuity from baseline to one year was not significantly different between the intervention and control participants.