

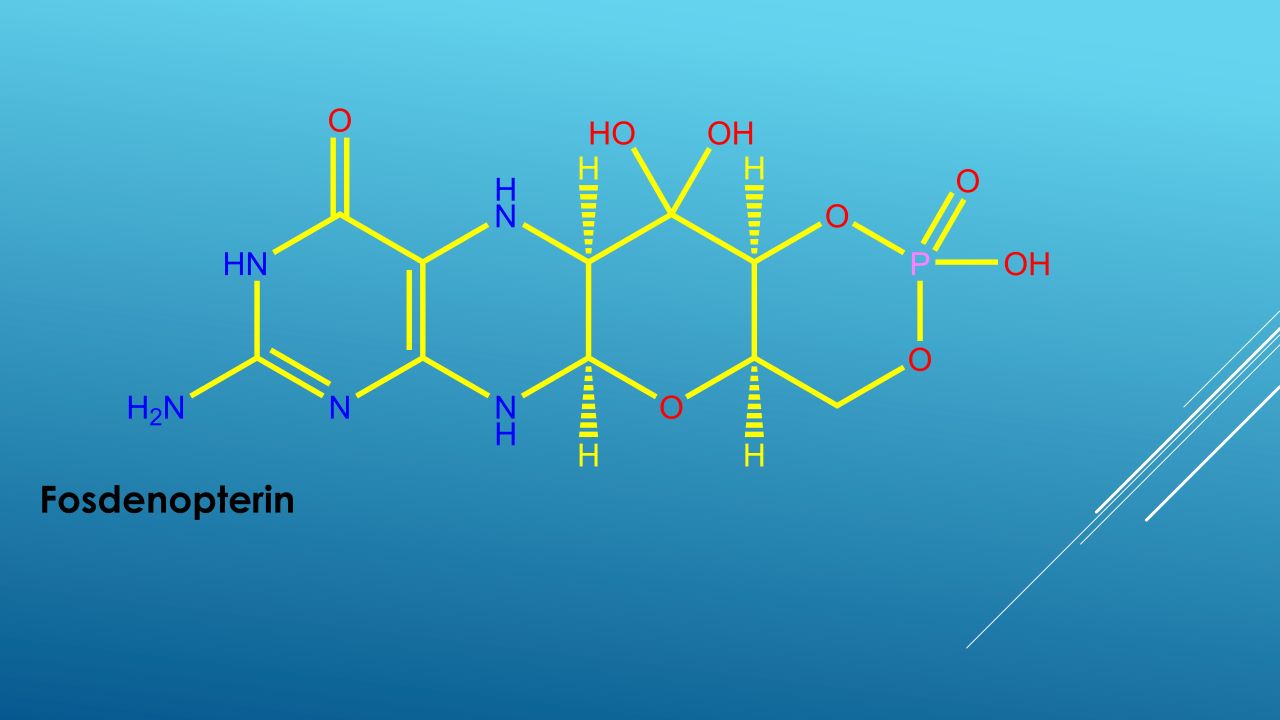
Nulibry™ (fosdenopterin) is the first and only US Food and Drug Administration (FDA) approved treatment indicated to reduce the risk of mortality in patients with molybdenum cofactor deficiency (MoCD) Type A.
Initially developed by Alexion Pharmaceuticals, the fosdenopterin (ALXN1101) was acquired by BridgeBio Pharma in the late stage of development in June 2018. BridgeBio Pharma subsequently launched a new subsidiary, Origin Biosciences, for the further clinical development and commercialisation of the therapy.
Recommended Buyers Guides
Nulibry is available in a single-dose vial containing 9.5mg of fosdenopterin, as a white to pale yellow lyophilised powder or cake for reconstitution for intravenous infusion.
Nulibry approvals
In December 2019, Origin Biosciences initiated a rolling submission of a new drug application (NDA) for fosdenopterin to the FDA for the treatment of patients with MoCD Type A. In September 2020, the FDA accepted the application and granted priority review designation to the drug.
Nulibry (fosdenopterin) secured FDA approval in February 2021. The drug also holds breakthrough therapy designation and rare paediatric disease designation in the US and orphan drug designation in the US and Europe.
MoCD Type A causes and symptoms
MoCD Type A is an ultra-rare, life-threatening genetic disease caused by a mutation in the molybdenum cofactor synthesis 1 gene (MOCS1). The mutation results in inadequate production of cyclic pyranopterin monophosphate (cPMP), which is essential to produce molybdenum cofactor (MoCo). Insufficient cPMP production eventually leads to MoCo deficiency.
MoCo is vital for the normal functioning of sulfite oxidase (SOX), an essential enzyme required to prevent the accumulation of highly toxic sulfite in the brain. The build-up of sulfite and secondary metabolites such as S-sulfocysteine (SSC) in the brain causes rapid, progressive and irreversible neurological damage.
MoCD Type A is an autosomal recessive, inborn error of metabolism that progresses rapidly and causes death in early childhood. The most frequent signs and symptoms of MoCD include feeding difficulties, encephalopathy, intractable seizures, high-pitched cries, exaggerated startle reactions, increased tightness of muscle tone (hypertonia) and reduced muscle tone (hypotonia).
Nulibry mechanism of action
Nulibry is a substrate replacement therapy that provides an exogenous source of cPMP. The cPMP is then converted into molybdopterin, followed by a further conversion into MoCo, which is crucial for molybdenum-dependent enzyme activities, including SOX.
Clinical trials on Nulibry
The FDA’s approval of Nulibry was based on the outcomes from three clinical studies that were compared to data from a natural history study in patients with MoCD Type A.
The first two studies were prospective, open-label, single-arm, dose-escalation studies, while the third was a retrospective, observational study.
A total of ten patients with confirmed MoCD Type A who received recombinant cPMP (rcPMP) were enrolled in the third study. Among the ten patients, six were later enrolled in the first study to receive Nulibry. The second study was conducted on one patient with MoCD Type A who had not been previously treated with rcPMP.
The efficacy of Nulibry in treating MoCD Type A was evaluated by the combined analysis of 13 treated patients from all the studies compared to 18 genotype-matched, untreated patients in the natural history control group.
The patients treated with Nulibry or rcPMP demonstrated an 82% reduction in the risk of death compared to the untreated group. The survival rate in patients treated with Nulibry or rcPMP was 84% at three years, compared with 55% in untreated, genotype-matched patients in the natural history control group.
The most common adverse reactions observed in Nulibry-treated patients were catheter-related complications, fever, viral infection, pneumonia, vomiting, coughing, viral upper respiratory infection, gastroenteritis, diarrhoea and inflammation in the ear, stomach and intestines.