

Rukobia (fostemsavir) is an antiretroviral drug, developed by ViiV Healthcare, for the treatment of HIV-1 infection in adult patients.
ViiV Healthcare is a joint venture between GlaxoSmithKline (GSK), Pfizer and Shionogi. Rukobia is intended to be taken with other antiretroviral drugs for the treatment of heavily treatment-experienced (HTE) multi-drug resistant HIV patients, who are failing the current antiretroviral regimen due to factors such as intolerance, resistance or other safety concerns.
Recommended Buyers Guides
The company filed a new drug application (NDA) with the US Food and Drug Administration (FDA) for fostemsavir in December 2019. A marketing authorisation application (MAA) for the drug was submitted to the European Medicines Agency (EMA) in January 2020. The FDA approved the drug in July 2020.
Rukobia is a novel attachment inhibitor available as beige, film-coated, oval, biconvex extended-release tablet, containing 600mg of fostemsavir for oral administration.
HIV causes and symptoms
Human immunodeficiency virus (HIV) is of two types- HIV-1 and HIV-2; HIV-1 is more virulent and infectious.
HIV destroys the CD4 T-cells in humans, causing HIV infection. The disease compromises the immune system of the individual and makes them susceptible to infections. It can easily be transmitted through infected blood and body fluids, which can result in AIDS, the late-stage of HIV infection, if not treated.
HIV infection causes flu-like symptoms such as fever, sore throat, muscle aches and fatigue. Approximately 1.2 million people in the US are currently living with HIV infection.
Rukobia mechanism of action
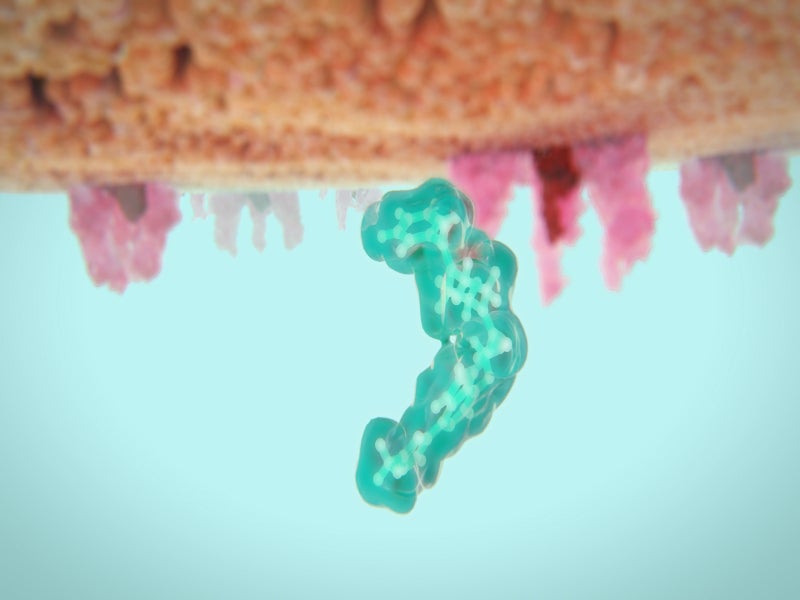
Rukobia is an HIV-1 gp120-directed attachment inhibitor. It is a prodrug of temsavir, a small-molecule HIV-1 attachment inhibitor.
After administration, Rukobia is hydrolysed to its active entity temsavir, which binds directly to the gp120 subunit present in the HIV-1 envelope glycoprotein gp160 on the surface of the virus. This blocks the interaction of the virus with the cellular CD receptors on the host immune system and prevents its attachment.
Temsavir can also block the other gp120-dependent processes after the attachment crucial for viral entry.
Clinical studies on Rukobia
FDA approval of Rukobia is based on the results of an international phase III, partially-randomised, double-blind, placebo-controlled clinical study, BRIGHTE.
The study enrolled 371 HTE HIV-1 infected patients with multidrug resistance; 272 patients were studied within the randomised cohort, while the remaining 99 patients were studied in a nonrandomised cohort.
The primary goal of the study was to evaluate the viral load decline in patients.
In the randomised cohort, patients were randomised to receive either 600mg Rukobia twice daily or placebo. On day 8, 65% of patients receiving Rukobia demonstrated a viral load reduction of more than 0.5 log10 copies per mL, while 46% of patients showed a reduction of greater than 1 log10 copies per mL in the patient’s blood.
Only 19% and 10% of patients receiving placebo showed a reduction in viral load from baseline of greater than 0.5 log10 copies per mL and greater than 1 log10 copies per mL, respectively.
In the randomised cohort, patients were also randomised to receive either Rukobia plus an optimised background therapy (OBT) or placebo. At week 26, 40% of patients who were administered with Rukobia plus OBT demonstrated a decline in viral load of greater than 40 copies per mL, whereas, at week 96, 30% of patients receiving Rukobia plus OBT showed a viral decline of more than 40 copies per mL.
In the nonrandomised cohort, approximately 37% of patients showed HIV-1 RNA <40 copies per mL at week 24 and week 96.
Nausea was the most common adverse event reported in the patients during the clinical trials.