

Ruzurgi® (amifampridine) is indicated for the treatment of adults and children aged six years and above with Lambert-Eaton myasthenic syndrome (LEMS), a rare autoimmune condition characterised by muscle weakness, which can be severe and even life-threatening.
Ruzurgi is an oval, functionally scored, white to off-white oral tablet available in 10mg strength. The drug was initially developed as a phosphate salt of amifampridine by Assistance Publique Hôpitaux de Paris.
Recommended Buyers Guides
Jacobus Pharmaceutical, a US-based pharmaceutical company, received the US Food and Drug Administration (FDA) approval for the manufacturing and commercialisation of Ruzurgi for the treatment of LEMS in patients aged six to 17 years in May 2019, following the submission of a new drug application (NDA) for amifampridine in June 2018 and receipt of a priority review designation.
In July 2022, American biopharmaceutical company Catalyst Pharmaceuticals (Catalyst) acquired certain intellectual property rights held by Jacobus, including the development and marketing rights of Ruzurgi in the US and Mexico.
In December 2019, Jacobus submitted a marketing application to Health Canada in collaboration with Medunik Canada to provide Ruzurgi to Canadian patients. Medunik received initial marketing authorisation for Ruzurgi by Health Canada in August 2020. The approval was initially suspended in June 2021 and then restored in the same month. It was then suspended again in March 2022 and reinstated once more in January 2023 based on a favourable Canadian federal court of appeal ruling.
ORSPEC Pharma obtained authorisation for Ruzurgi from the Therapeutic Goods Administration (TGA) Australia, for the treatment of LEMS in adults and children aged six years and above, in September 2021.
LEMS causes and symptoms
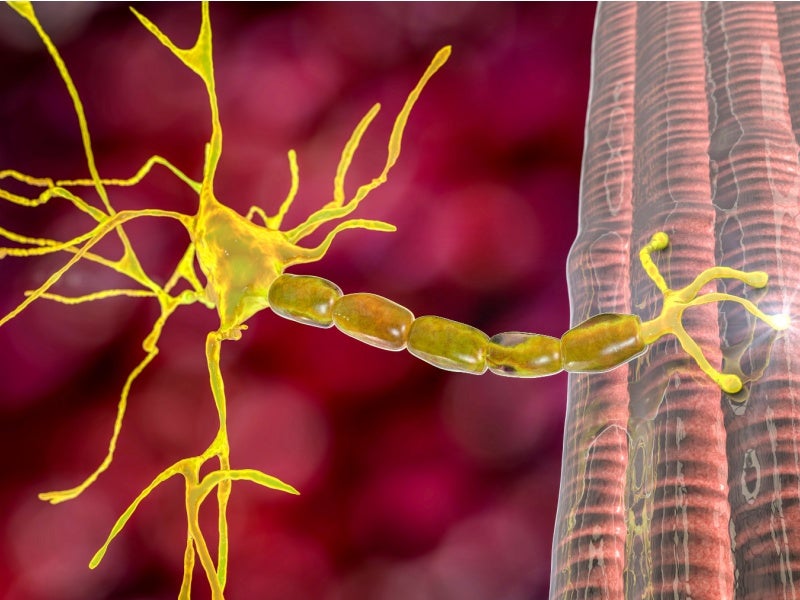
LEMS is a rare autoimmune disease that attacks the neuromuscular junctions of the body. It destroys the nerve cells’ ability to send signals to the muscle cells. LEMS is named after two neurologists Edward Lambert and Lee Eaton, who described the disease.
LEMS is a small molecule potassium channel blocker that hinders the calcium channels on nerve endings that are responsible for releasing acetylcholine, a chemical messenger that generates muscle contraction. Reduced levels of acetylcholine cause improper muscle contraction, weakening the muscles.
Patients suffering from cancers are susceptible to the condition, which can affect individuals of any age. Three in one million people worldwide are affected.
Some of the common symptoms associated with the disease are weakness in the legs, hip, upper arms, eye muscles and shoulders, as well as difficulty walking and talking.
Ruzurgi’s mechanism of action
Amifampridine is a potassium channel blocker with a broad spectrum of action. It is a quaternary ammonium drug that elevates presynaptic calcium concentrations and prolongs the action potential by blocking presynaptic potassium channels. The exact mechanism by which amifampridine works as a treatment for LEMS patients is yet unclear.
Clinical trials on Ruzurgi
The FDA’s approval of the drug was based on the positive results of a Phase II, randomised, double-blind, placebo-controlled, withdrawal study named DAPPER. The study enrolled 32 adult patients who were already receiving Ruzurgi for three months before being enrolled in the clinical study.
Patients continuing with Ruzurgi were compared with those who switched to a placebo upon beginning the trial. The primary endpoint of the study was the degree of change in the Triple Timed Up and Go test (3TUG) after the withdrawal of the drug.
3TUG is the amount of time taken by patients to rise from a chair, walk for three minutes and return to the chair thrice consecutively. Higher scores suggest greater deterioration.
Patients administered with Ruzurgi did not witness more than 30% deterioration in the final round of the 3TUG test while 72% of patients randomised to placebo experienced more than 30% deterioration in the final 3TUG test.
The most common adverse events reported during the clinical trial were burning or prickling sensations, abdominal pain, dyspepsia, dizziness, and nausea.
Controversy associated with amifampridine approval
Catalyst contested the FDA’s approval of Ruzurgi in June 2019, asserting that it infringed upon its statutory rights, leading to legal action against the FDA and several affiliated parties. It challenged the approval and associated drug labelling. Additionally, Catalyst claimed that the approval contravened its statutory entitlements to Orphan Drug Exclusivity and New Chemical Entity Exclusivity under the Federal Food, Drug, and Cosmetic Act (FDCA).
Catalyst obtained exclusive rights for Firdapse®, which contains amifampridine phosphate, for the North American market in October 2012. Firdapse had FDA orphan drug designation for LEMS and breakthrough therapy designation for LEMS in August 2013. Firdapse also received orphan drug designation for Myasthenia Gravis (MG).
The FDA approved Firdapse to treat LEMS in adults in November 2018. The drug is also approved in Canada for LEMS in adults. BioMarin Pharmaceutical markets Firdapse in the European Union for LEMS treatment in adults aged 18 and above, following its approval in April 2010.
In February 2022, the District Court ruled in favour of Catalyst, invalidating the FDA marketing approval for Ruzurgi.
Catalyst settled patent infringement litigation with Jacobus Pharmaceutical and PanTHERx Rare in July 2022, acquiring rights to develop and commercialise Ruzurgi in the US and Mexico.