Alpha-1-antitrypsin (A1AT) is a glycoprotein that protects connective tissues from proteolytic damage inflicted by serine proteases. This occurs when neutrophils secrete serine proteases to destroy invading bacteria following an inflammatory stimulus, but their onslaught does not differentiate between invading and host cells so requires regulation. A subset of the population has an uncurable genetic disease known as alpha-1-antitrypsin deficiency (A1ATD), which can result in dysfunctional A1AT variants incapable of inhibiting serine proteases. Consequently, A1ATD patients can suffer from complications following tissue damage such as chronic obtrusive pulmonary disease (COPD) or liver fibrosis/cirrhosis.
Liver damage in A1ATD patients manifests because disease-causing alleles promote A1AT polymerisation, producing chains of ordered polymers. Although some polymers will be degraded by intracellular quality control mechanisms, others will accumulate in the liver, causing hepatic damage. This results in insufficient levels of circulating A1AT due to inadequate excretion from the liver, leaving the lungs susceptible to potent serine protease attacks that can trigger COPD. Some patients with A1ATD-related COPD benefit from augmentation therapy, which supplements A1AT levels using donor plasma, but this treatment is not approved in most high incidence geographies such as the UK. As there is minimal treatment and no cure, A1ATD patients’ symptoms such as shortness of breath, wheezing, chest pains and increased susceptibility to chest infections become progressively worse as the disease advances.
Go deeper with GlobalData

Access deeper industry intelligence
Experience unmatched clarity with a single platform that combines unique data, AI, and human expertise.
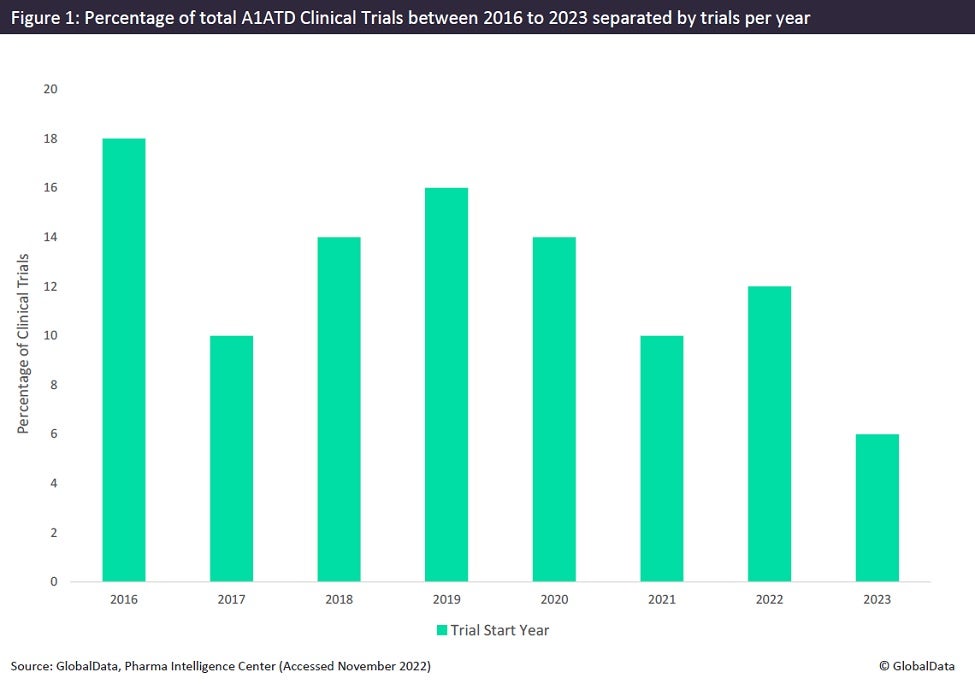
Although A1ATD is classified as a rare disease, it is commonly considered to be a prevalent disease, rarely diagnosed. Studies have demonstrated that targeted screening approaches show the frequency of A1ATD associated alleles to be four times higher in a randomly selected population than reported figures for that country. Significant factors contributing to this underdiagnosis include the multi-step algorithm requiring specialised equipment in a pathology laboratory to diagnose most disease-causing alleles, the generic nature of A1ATD symptoms leading to misdiagnosis, and the lack of A1ATD awareness in the general population and amongst medical practitioners. Figures derived from GlobalData demonstrate that the number of clinical trials concerning A1ATD has not entered double digits in recent decades and has steadily declined since 2016 (Figure 1).
Longitudinal clinical trials concerning A1ATD are not currently feasible because the available method of measuring the quantity of aggregated polymers in the liver using invasive liver biopsies is not ethically viable in the long term. As polymers are the pathological hallmark of A1ATD and correlate to the severity of the disease, this is a major setback for A1ATD research and significantly impacts the amount of A1ATD clinical trials initiated. Luckily, this has not completely discouraged research into A1ATD, but it was highlighted by Takeda Pharmaceuticals when reporting promising results from their Phase II trial for their drug Fazirsiran. This drug intends to eliminate the polymer burden in A1ATD sufferers by preventing the expression of mutant A1AT. Hopefully, this success can continue in the Phase III trials estimated to begin in March next year; if so, a drug that satisfies the largely unmet clinical needs of A1ATD patients may be on the horizon.