
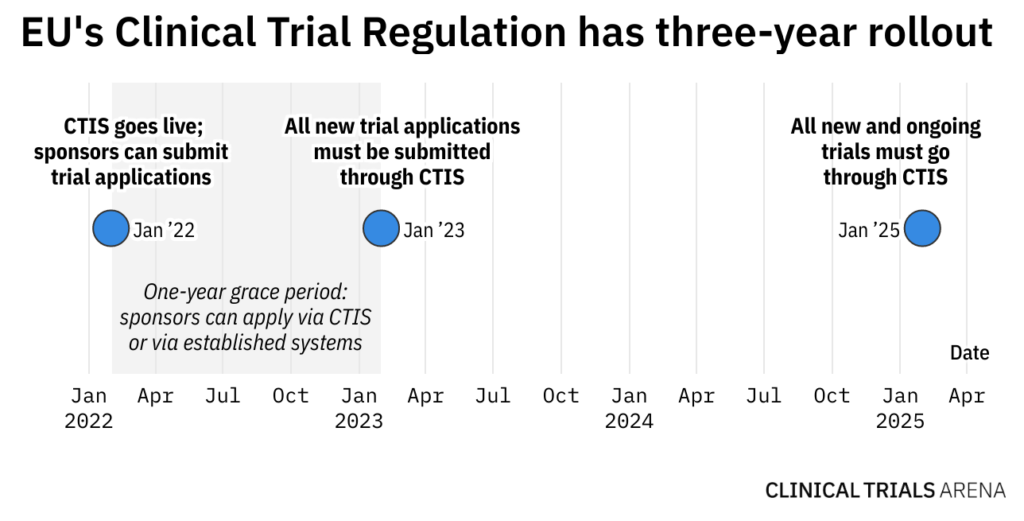
The EU’s Clinical Trials Regulation (CTR) and its corresponding portal Clinical Trials Information System (CTIS) went live at the end of January. Many see it as valuable tool to streamline and improve clinical trial regulatory and ethics assessments in Europe. Yet, almost six months later, its target users are yet to jump in.
According to the EMA’s June 23 key performance indicator (KPI) report, 92 clinical trial applications were submitted via CTIS since February, eight of which were authorised. In contrast, 758 applications were uploaded by individual member states via the traditional pathway EudraCT. Of these, 165 were authorised but this number is expected to increase.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
It was not unexpected that CTIS would have some technical hiccups, with the EMA being proactive to address these issues. Difficulties range from the need to upload documents using a specific format, to challenges in submitting applications for trials with a complex design. Questions swirl if the agency’s decision to use an online portal approach, which needs manual data input, instead of a system that can synchronise with an internal one, was the right choice.
It was also not unexpected that CTIS would have slow early uptake among sponsors, owing to many of them planning to submit their new trials via the old system anyway. There were nine applications via CTIS in February, rising to 36 in May. Ongoing trials don’t have to go through the new system yet and doing so now may add unnecessary paperwork.
Yet, from January 2023, all new submissions must go through CTIS, and this deadline is fast approaching. With sponsors yet to fully embrace the new system, they might need to act fast to get over the necessary learning curve to make the most of the portal. Not doing so can risk application errors and could have a domino effect on the trial’s start date.
CTIS tech issues surface
On July 1, the EMA held a webinar discussing CTIS progress thus far, with users able to share their experiences. There were minor technical issues raised such as the need of a PDF version of a document instead of one saved in Word.
There were more complex issues: an Oslo University Hospital representative shared that CTIS is not optimal for platform trials as each new arm had to be entered as a new trial. This leads to full fees for each arm in many countries instead of update fees. Also, the same documents are filed multiple times, and are assessed individually. The EMA stated at the event that it prioritises operational stability and support of portal users and will address feedback raised during the webinar.
Another CTIS issue is that clinical trial sites and vendors need to be registered in an organisation management service (OMS) to enable them to use CTIS, says Sarah Bly, Worldwide Clinical Trials regulatory intelligence associate director. There are issues regarding how OMS and CTIS are connected, disallowing certain vendors to be added to the CTIS vendor database, she tells Clinical Trials Arena. OMS manages substance, product, organisation and referential (SPOR) master data domains in the pharmaceutical regulatory processes.
But Bly notes that overall, the new system is user-friendly. “There is a good help desk at the EMA—it is continuously ramped up with more resources.”
CTIS portal approach has caveats
Was it a good idea to have CTIS be an online portal? Phlexglobal chief product officer Jim Nichols says probably not. “The fact that EMA went with this portal approach versus data transmission from system to system is in some ways disappointing because it felt like we really didn’t make an advancement.” The CTIS is a manual process of going into a portal and typing in data with no synchronisation with internal systems, he explains.
Manual data entry can create risk and inefficiency, Nichols adds. “If we are moving into a world of transmitting and moving data from point A to point B, we need to be confident that the data in one system is the same in another without manual steps in the middle where somebody could make a mistake,” he notes. Sponsors will have to figure out a tracking tool to avoid such issues, he says.
Uptake expected to rise
Slow initial CTIS uptake is not surprising. “We have not seen a flurry of submissions under the new directive yet, and I think it is understandable with something that represents quite a substantial management change,” says Paul Bridges, Parexel executive vice president of regulatory and access.
Companies interpret 2022 as the year of last submission under the old directive rather than making the first submission through CTIS, Bridges says. “Companies don’t want to necessarily be the pilots in what is a highly regulated environment,” he adds.
Sponsors with ongoing trials have little incentive to try CTIS just yet. Bone Therapeutics CEO Miguel Forte says the company has no experience with the new system. Its ongoing investigations were approved prior to CTIS, and it made sense for trial amendments to be via existing pathways.
“My objective as a sponsor is getting my study approved as quickly as possible using the materials that I have already prepared,” Forte adds. The final milestone for the CTIS rollout is on January 31, 2025, when all new and ongoing clinical trials will have to be managed through CTIS.
How to get ready
Sponsors should start preparing now ahead of the next deadline. They can do so by starting conversations with regulatory experts and relevant authorities, says Aman Khera, Worldwide Clinical Trials global head of regulatory strategy. “If there is an opportunity to engage with a regulatory agency or the EMA, you should be doing it. Now it is not the time to be shy, but to take advantage of it,” Khera adds.
Pre-application advice will ensure that risks of application lapses and withdrawals are avoided as filing process timelines are more rigid, Bly adds. Now, member states have 60 days to evaluate the initial application, in contrast to before when there was a lack of clarity, causing each country to have their own timelines. According to the KPI report, since the CTIS launched, eight submissions were withdrawn by the sponsors and six lapsed at the time of validation. Currently, 69 clinical trial applications are still under evaluation.
Sponsors need to focus on the quality of the documents and ensure they are submission-ready, Khera says. A clinical trial application process has two parts: Part I contains documents that feature scientific and medical product documents, while Part II has national and patient-level documents. Both parts need to be ready at the same time when applying on CTIS, Khera says.
Sponsors should decide who oversees documents internally, Nichols says. “It is important to map out what and from where documents are needed and what is the chain of custody around the information,” he adds. Sponsors should identify a single point of responsibility, a clinical or regulatory team member who has gone through training and understands the submission process, while other parts of the organisation should be aware of their dependency and tasks.
To have first-hand experience, sponsors need to identify which studies would benefit from being uploaded via the new system. Uploading trials with a simple design is ideal as a starter because complex trials with many sites or in indications with hard-to-recruit patients can cause extra amendments during the submission process, Bridges explains.
And, as for those who have already been through the CTIS process, they should share their experiences with other sponsors. The system will be improved by the industry having such conversations, instead of doing structured training, Forte says. “We need to make our part in the support of the system by being keen to use it and make sure we adopt everything.”
Takeaways:
- CTIS technical issues are not unexpected and so users need to be vocal on their experience for challenges to be addressed by the EMA.
- Sponsors with an eye to initiate clinical trials in the future will need to start adjusting to use the new system as all new applications will have to go through CTIS from January 31, 2023.
- Transition to the new system can be made easier by opening lines of communication with regulatory agencies and ensuring all required documents are ready and fit for the platform.
