



Lodotra was developed utilising SkyePharma’s proprietary GeoClock and GeoMatrix technologies, for which Horizon Pharma holds an exclusive worldwide license for the delivery of corticosteroids. The drug was developed jointly with Nitec Pharma, which was formed in 2004 as a spin-out from Merck KGaA. Nitec merged with Horizon Pharma in April 2010.
The Marketing Authorisation Application was submitted to the European Medicines Agency (EMA) in 2006. The drug was approved for the treatment of RA patients with associated morning stiffness in Europe in March 2009.
Recommended Buyers Guides
It is currently approved in 16 European countries, the US and Israel. Lodotra is being marketed by Mundipharma in Europe. Mundipharma also has rights to the product for certain Asian countries and South Africa.
Horizon Pharma submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) in September 2011. The trade name for Lodotra in the United States is Rayos. In July 2012, Rayos was approved by the FDA for the treatment of many diseases including rheumatoid arthritis. It was launched in the US market in December 2012.
A Phase III clinical trial of Lodotra for the treatment of polymyalgia rheumatica (PMR) is being planned. This indication is not included in the NDA.
Rheumatoid Arthritis
Rheumatoid Arthritis is a chronic illness that causes inflammation of joints and surrounding tissues. It is an autoimmune disease in which the immune system attacks healthy body tissue.
The exact cause of RA remains unknown. Infection, genes or hormone changes may cause the disease. RA is more common in women than in men and it can occur at any age.
The disease progresses slowly and affects the joints of the body equally on both the sides. The most commonly impacted parts are wrist, fingers, feet, knees and ankles.
The initial symptoms include minor joint pain, stiffness and fatigue. Other symptoms include morning stiffness, joint pains, deformation of the joints, chest pain, dry eyes / mouth and a numbness / tingling sensation in hands and feet.
It is estimated that 1.8 million people in the U.S suffer from RA.
Lodotra – drug mechanism of action
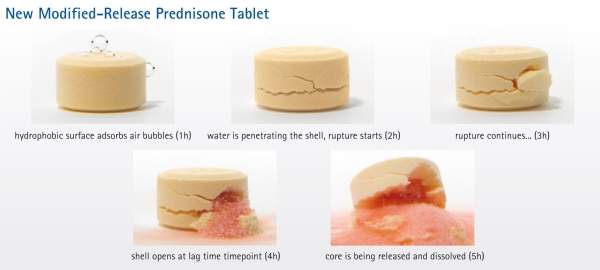
Lodotra is comprised of an active core containing prednisone, which is encapsulated by an inactive porous shell. The inactive shell acts as a barrier between the product’s active core and a patient’s GI fluids.
Lodotra is intended to be administered at bedtime. At approximately four hours following bedtime administration of the drug, water in the digestive tract diffuses through the shell, causing the active core to expand, which leads to a weakening and breakage of the shell and allows the release of prednisone from the active core.
The enzymes produced by the liver convert prednisone into prednisolone. Prednisolone has anti-inflammatory and anti-immunosuppressive characteristics.
It inhibits high levels of pro-inflammatory cytokine when the patient is asleep and is thought to reduce early morning joint stiffness.
Pre-clinical studies of the treatment medication
Pre-clinical trials were conducted in rats to evaluate the safety of the drug. Rats were administered 33mg/kg of drug for seven to 14 days.
Histotoxic effects (myonecrosis) were observed on administration of 0.5 to 5mg/kg of drug in guinea pigs and 4mg/kg dosage in dogs.
Clinical trials of Horizon Pharma’s rheumatoid arthritis drug
Horizon Pharma completed two pivotal Phase III clinical trials evaluating Lodotra for the treatment of RA. The Circadian Administration of Prednisone in Rheumatoid Arthritis-1, or CAPRA-1 trial, investigating the efficacy of Lodotra in the treatment of RA, was designed to support MAA approval in Europe.
The second pivotal Phase III clinical trial, Circadian Administration of Prednisone in Rheumatoid Arthritis-2, or CAPRA-2 trial, was designed to support an NDA submission for US marketing approval. The drug was approved by the US FDA based on the data received from CAPRA- 1 and 2 clinical studies.
CAPRA- 1 (circadian administration of prednisone in RA) was conducted to determine the safety and efficacy of the drug in the treatment of RA. The 12 week, double-blind, placebo-controlled study in Europe evaluated 288 RA patients. CAPRA-1 compared the night-time administration of Lodotra with the morning administration of immediate release prednisone at the same individual dose (average dose of 6.7 mg). Results from CAPRA-1 demonstrated the following:
– A statistically significant reduction in morning stiffness compared to patients in the immediate release group was the primary outcome of the trial (22.7% for Lodotra compared to 0.4% for immediate release prednisone (p-value = 0.045)).
– Patients treated with Lodotra had a reduction in IL-6 levels of approximately 29% (relative median change), which was statistically significant, while corresponding IL-6 levels following treatment with immediate release prednisone remained constant.
The most commonly reported treatment-emergent adverse events were a flare in RA-related symptoms (7.6% for Lodotra compared to 9.0% for immediate release prednisone), abdominal pain (3.5% for Lodotra compared to 5.6% for immediate release prednisone), nasopharyngitis (2.8% for Lodotra compared to 5.6% for immediate release prednisone), headache (4.2% for Lodotra compared to 2.8% for immediate release prednisone) and flushing (2.8% for Lodotra and 4.2% for immediate release prednisone).
Nine month extension study
Following the 12-week CAPRA-1 study, patients were followed in a nine-month, open-label extension study, which included 249 RA patients, 219 of whom completed the extension study. Results showed that patients who continued treatment with Lodotra experienced a 55% reduction in the duration of morning stiffness. Furthermore, patients newly assigned to Lodotra exhibited a 45% reduction in the duration of morning stiffness over the nine-month course of this extension study. These patients also experienced a 50% median reduction in IL-6 levels that also corresponded to improvements in the duration of morning stiffness following daily administration of Lodotra.
The most commonly reported treatment-emergent adverse events in the extension study were a flare in RA-related symptoms (14.5%), flushing (5.2%), upper respiratory tract infections (2.8%), back pain (2.8%) and weight increase (2.8%).
Adverse events indicative of aggravated hypothalamic-pituitary adrenal, or HPA, axis suppression, typical of high dose prednisone administration, were not observed.
CAPRA-2 trial details
CAPRA-2 trial enrolled around 350 RA patients with inadequate response to DMARD therapy. The trial was conducted in US patients to evaluate the safety and efficacy of the drug in comparison with placebo.
The trial was initiated in March 2008 and completed by July 2009. Patients were administered either 5mg Lodotra or were given the placebo.
Results showed patients treated with Lodotra experienced a statistically significant improvement in ACR20 response criteria compared to patients in the placebo group (48.5% vs. 28.6%; p-value = 0.0002), which met the primary endpoint. In addition, patients taking Lodotra experienced a statistically significant improvement in the more stringent ACR50, response criteria (22.7% vs. 9.2%; p-value = 0.0027) and ACR70, response criteria (7.0% vs. 2.5%; p-value = 0.0955).
Patients treated with Lodotra also experienced a statistically significant reduction in morning stiffness compared to patients in the placebo group (56.5% vs. 33.3%; p-value = 0.0008).
In this study, the most commonly reported treatment-emergent adverse events were joint pain (10.4% for Lodotra compared to 20.2% for placebo), RA flare (6.5% for Lodotra compared to 9.2% for placebo), nasopharyngitis (4.8% for Lodotra compared to 3.4% for placebo) and headache (3.9% for Lodotra compared to 4.2% for placebo).
Phase III clinical trials of Horizon Pharma’s Lodotra
A Phase III clinical trial of Lodotra for the treatment of polymyalgia rheumatica (PMR) is being planned. In an investigator-initiated study, recently presented at the American College of Rheumatology, ten PMR patients given either Lodotra (MR prednisone) at night or prednisone in the morning.
Results showed that IL-6 and symptoms of morning stiffness were both suppressed more by nighttime low dose MR prednisone, which may be because PMR, like RA, has a marked circadian variation in serum IL-6. This observation raises the possibility that PMR may be controlled by lower doses of MR prednisone given at night compared to current conventional morning treatment.