
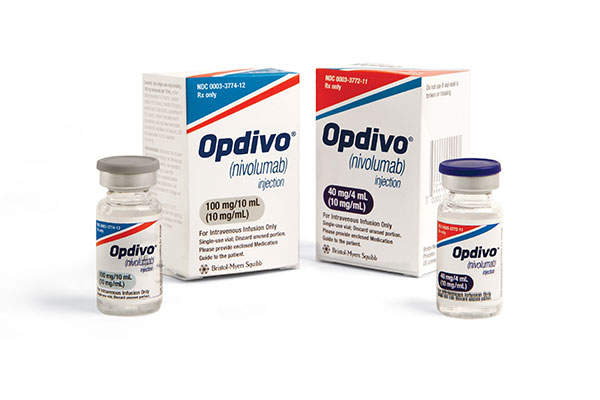
Opdivo (nivolumab) is indicated for the treatment of unresectable or metastatic melanoma in patients with a positive BRAF V600 mutation who were previously treated and made progression with Yervoy (ipilimumab).
Recommended Buyers Guides
The drug was discovered and developed by Bristol-Myers Squibb (BMS). The company partnered with Japanese firm Ono Pharmaceutical in July 2014 to develop and commercialise Opdivo, as well as several other immuno-oncology assets in Japan, South Korea and Taiwan.
The US Food and Drug Administration (FDA) accepted a biologics license application (BLA) of Opdivo for priority review. The drug was also granted breakthrough therapy designation in September 2014.
BMS received approval from the FDA in December 2014, three months ahead of the final Prescription Drug User Fee Act (PDUFA) date. The drug was also approved as adjuvant therapy in patients with completely resected melanoma in December 2017.
The European Medicines Agency (EMA) validated the marketing authorisation application (MAA) of Opdivo in Europe. The Committee for Medicinal Products for Human Use (CHMP) granted accelerated assessment in September 2014. In June 2017, the drug received approval by the European Commission (EC).
Opdivo received FDA approval to treat previously untreated patients at intermediate or poor-risk of advanced renal cell carcinoma in combination with Yervoy in April 2018.
The FDA also approved Opdivo for the treatment of patients with advanced small cell lung cancer (SCLC) in August 2018.
The drug has also received FDA approval for the treatment of Hodgkin lymphoma, recurrent or metastatic squamous cell carcinoma of the head and neck and advanced urothelial carcinoma.
The Opdivo and Yervoy combination also received approval in Taiwan for first-line treatment of intermediate or poor-risk advanced renal cell carcinoma in November 2018.
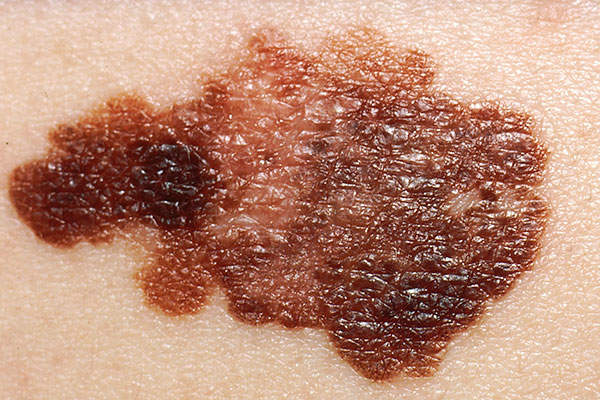
Metastatic melanoma, the deadliest form of skin cancer
Melanoma is a type of skin cancer that occurs due to the overproduction of melanocytes in the skin. In metastatic melanoma, cancer has spread to other parts of the body, such as the lungs, brain and lymph nodes. Survival rate decreases in detection at later stages.
Opdivo’s mechanism of action against melanoma
Opdivo contains an active component called nivolumab, which is a human immunoglobulin G4 (IgG4) monoclonal antibody.
The intravenously administered drug inhibits the proliferation of T-cells and the production of cytokine by binding to the PD-1 receptor, which blocks its interaction with PD-L1 and PD-L2 and decreases tumour growth.
Clinical trials on Opdivo
Yervoy is the first drug approved for the treatment of metastatic melanoma in the US.
The FDA’s approval for Opdivo was based on tumour response rate and durability response. Evaluation of the drug’s safety and efficacy took place over two Phase III clinical trials, which enrolled advanced melanoma patients previously treated with BRAF inhibitor Yervoy.
The first Phase III study evaluated Opdivo’s efficacy in a single-arm, non-comparative planned interim study, which enrolled 120 patients.
Patients received a dose of 3mg / kg Opdivo intravenously over 60 minutes every two weeks, with follow-up within a minimum of six months. The study was followed up by a Phase III CheckMate-037 trial for six months.
Results showed that patients administered with Opdivo achieved a 32% response rate. Approximately 3% of patients (4/120) achieved a complete response and 28% (34/120) achieved a partial response.
Out of 38 subjects, 87% (33 patients) had continuous responses from 2.6 months to ten months, including 13 patients with continuous responses for six months or longer. Responses were also observed in patients with and without BRAF mutation.
The EC’s approval of Opdivo was based on results from a Phase II clinical trial named CheckMate-275, an open-label, single-arm, multi-centre study evaluating Opdivo in patients with locally advanced or Metastatic Urothelial Carcinoma (mUC).
The study enrolled 270 patients who were administered with Opdivo 3mg / kg intravenously every two weeks until disease progression or unacceptable toxicity. The primary endpoint of the study was objective response rate (ORR) and the secondary endpoints included progression-free survival (PFS) and overall survival (OS).
Results from the study demonstrated that approximately 20% of patients administered with Opdivo responded to the treatment. The study also found that the percentage of patients with a complete response was 3% and those with a partial response was 17%.
The safety of Opdivo was evaluated in a randomised, multinational, multicentre and open-label Phase III clinical trial known as CheckMate-037. The study was conducted in 90 centres across 14 countries, enrolling patients with previously treated advanced melanoma, including 76% M1C patients, 18% with a history of brain metastases and 56% with elevated lactate dehydrogenase (LDH) levels.
Patients were treated with either Opdivo 3mg / kg every two weeks or chemotherapy, which included either single-agent dacarbazine at 1,000mg / m2 administered every three weeks or a combination of carboplatin AUC 6 every three weeks plus paclitaxel 175mg / m2 every three weeks.
The preliminary objective of the study was to check the objective response rate (ORR). The study is still ongoing to determine the overall survival benefit.
The FDA’s approval of Opdivo for renal cell carcinoma was based on positive results of phase three clinical study CheckMate -214. Approval for SCLC was based on positive overall response rate and duration of response achieved in phase I / II clinical study CheckMate -032.
Opdivo clinical trial failures
Phase three trial CheckMate -548, which compared Opdivo in combination with temozolomide and radiation therapy to the standard of care, failed to meet one of its primary endpoints. It followed the failure of Phase III CheckMate -498 trial to meet its primary endpoint.
The CheckMate -498 trial compared Opdivo in combination with radiation to standard of care (temozolomide plus radiation) in newly diagnosed O6-methylguanine-DNA methyltransferase-unmethylated glioblastoma multiforme.
Side effects
The most severe adverse reactions reported in Opdivo-administered patients in the study include abdominal pain, hyponatremia, increased aspartate aminotransferase, and increased lipase.