
Need to know:


- EU’s Clinical Trials Regulation goes live on Monday (31 January). Its foundational Clinical Trials Information System (CTIS) is being presented as a one-stop shop for clinical trial applications, regulatory review, and public access to trial information.
- For the first year of rollout, stakeholders can use CTIS, but existing application frameworks will still exist. From January 2023, all new clinical trial applications will go through CTIS. Beginning 2025, all new and ongoing trials must go through the platform.
- Harmonisation is welcome, but processes within the Clinical Trials Regulation could introduce trial initiation delays. Sponsors should be diligent in choosing countries to which they apply.
- While CTIS allows for streamlining applications, issues could stifle its uptake momentum. For instance, sponsors with clinical trial application dossiers made with previous systems in mind are unlikely to be early CTIS adaptors.
It’s all systems go: EU’s Clinical Trials Regulation (Regulation (EU) No 536/2014) is launching on Monday (31 January). Its underlying clinical portal and database – Clinical Trials Information System (CTIS) – goes live on the same day. Both are envisioned to streamline clinical trial application, review, and supervision, and bolster transparency.
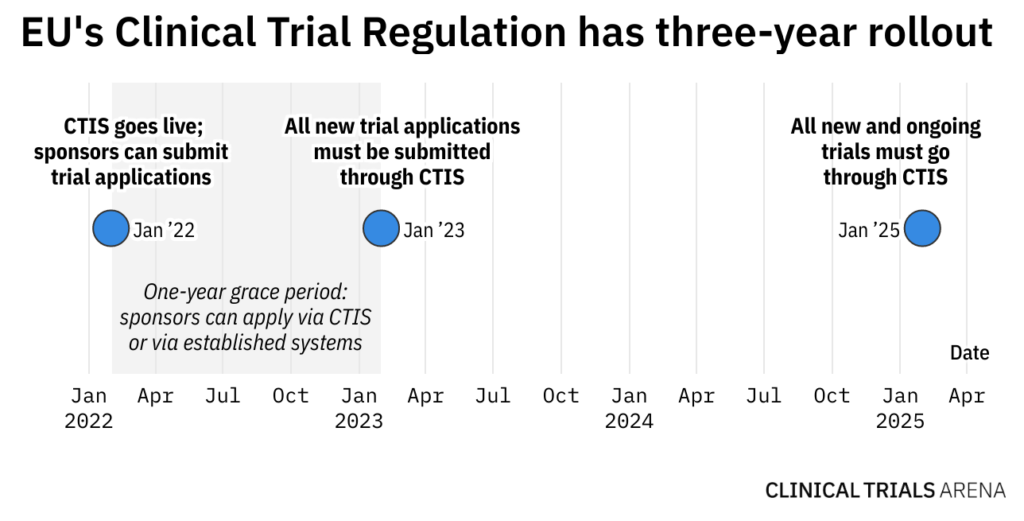
With CTIS, clinical trial sponsors can submit all regulatory and ethics assessments under a single application instead of applying to each EU member state individually. Industry and academic sponsors will have a one-year grace period before they must submit all new clinical trial applications through this system. Starting January 2025, all new and ongoing trials must go through CTIS.
CTIS represents great progress towards aggregating information and promoting trial execution in Europe, notes Bone Therapeutics CEO Miguel Forte. But the actual implementation could face early hiccups as sponsors and member states gain comfort with the new platform, he says. Sponsors will need to be strategic to avoid delays in initiating clinical trials, GCP Central founder and CEO Marieke Meulemans adds.
Advantages for multinational trials
The Clinical Trials Regulation and CTIS will make it easier to run multinational trials in Europe and expand existing trials into additional member states, said the chairperson of the Heads of Medicines Agencies (HMA) Management Group Karl Broich during a 25 January press conference. The HMA works with the European Medicines Agency (EMA) and the European Commission for the operational activities in the European medicines regulatory network.
This integration will prove particularly useful for running trials for rare diseases where patient populations are typically spread out among numerous member states, Broich said. CTIS could also facilitate a wider academic research infrastructure, as research will be less fragmented by country, added Fergus Sweeney, head of the EMA’s Clinical Studies and Manufacturing Task Force.
Overall, Europe has struggled to compete with the US and China in terms of running clinical trials and getting innovations to patients, Forte says. But an integrated, universal database could prompt the EMA to take steps toward increasing clinical trials in the continent, he adds.
As stated by EMA Executive Director Emer Cooke during the 25 January press conference: “CTIS is an enormous opportunity to put Europe back on the map as a world-leading research environment”.
Member states still independent
But while CTIS makes clinical trial submissions more straightforward, it doesn’t remove the power of individual stakeholders to make decisions, Forte told Clinical Trials Arena. While member states collaborate on Part I of the review process, they will still be independent when it comes to deciding if a certain trial can be staged in their respective countries, Meulemans explains.
Part I of an application features scientific and medical product documents while Part II contains national and patient-level documentation. Under the new system, member states will still authorise and oversee clinical trials, while the EMA manages the database and content publication.
Trial initiation delays?
Implementing the Clinical Trials Regulation could risk delaying the activation of clinical trial sites, notes Meulemans, who was a member of the CTIS Training Expert group of EMA for validation of the CTIS training material. The next two years will tell whether companies experience such delays, particularly for globally organised trials, she adds.
As an example, sponsors or academia can apply to run their trials in all countries at the same time. But the risk here is that each of these countries will have to provide input on Part I of the initial application, which requires a coordinated effort among countries, to be managed by a reporting member state, Meulemans notes.
Member states may have different opinions and views on the Part I documents that may not be seen as a critical issue to the reporting member state, Meulemans notes. But such opinions and views could still lead to unnecessary rejections or a trial approval but under certain conditions.
Sponsors may need to answer a long list of follow-up questions within a very short timeframe, and submit updated documents, Meulemans notes. The initial review runs for 60 days excluding the request for information period. Sponsors have a maximum of 12 calendar days to reply to any additional queries, she adds.
Alternatively, sponsors or academia can submit the Part I and II applications in targeted countries only. But the challenge here is that the trial may subsequently require additional sites in a new set of countries once the initial application is complete, Meulemans notes. There are alternative submission strategies that can be considered: such as the submission of Part I and II in one country only, or to delay the submission of Part II for specific member states that were already included in the Part I process.
While the timeline for a substantial amendment for the “addition of a member state” application is shorter than the main application (52 calendar days excluding request for information), it could still lead to delays, Meulemans notes. No parallel submissions are allowed in the Clinical Trials Regulation, meaning that no member state can be added until the initial submission process has been completed by all member states concerned. That said, the added member states cannot change the decision on Part I that was decided in the initial application, she states.
Portal learning curve likely
A significant learning curve with the CTIS is likely as sponsors and regulators transition to the new platform, says Forte. The evolving landscape of clinical trial evaluation and supervision could make implementation of this new IT system particularly challenging, Broich said at the briefing.
Most sponsors will welcome CTIS, but it could prove challenging for some small academic sponsors, Forte says. CTIS is a great system in principle, but more challenges could arise during its actual implementation, he notes.
For a successful implementation of CTIS, management of the first six months will prove key, according to Forte. Trial sponsors who already have existing applications with existing systems in mind will unlikely revise applications, he said. But, at around six months, applications that would be sent at that point will likely be sculpted for CTIS, he said. The EMA did not respond to a comment request.
